Does Cigarette Smoke Cause Interleukin 1 - Beta (Il-1ß) Production in Cardiac Stem Cells?
*Corresponding Author(s):
Wasana SumanasekeraDepartment Of Cell Biology, Sullivan University College Of Pharmacy, Louisville, United States
Tel:+1 5024138954,
Email:wsumanasekera@sullivan.edu
Abstract
Cigarette smoking leads to vast array of illnesses including cardiac diseases. The focus of our laboratory is to investigate the influence of cigarette smoke exposure on Cardiac Stem Cell (CSC) functions and study underlying mechanisms. Cardiac stem cell therapy is one of the emerging treatment options to treat myocardial damage. Therefore, studying cigarette smoke-induced modulations of CSC functions and responsible mechanisms are of furthest importance. The current study investigated whether CSCs produce interleukin 1 beta (IL-1ß), which is a potent pro-inflammatory cytokine, in response to exposure to cigarette smoke, with the view of understanding the role of interleukins as one of the causative agents in cigarette smoke- induced modulation of cardiac stem cell functions. Cigarette Smoke Extracts (CSE) were created by combusting camel filter-free cigarettes and dissolving the smoke puff in cell culture media based on a published method. No smoke control (0% CSE) and three different concentrations of CSE, which are 2%, 5%, and 10%, were utilized. CSCs were cultured and treated with CSE for 15 minutes, 1 hour, and 24 hours. Then from each treatment group media on top of the CSCs (supernatant) were removed and protein samples were quantified using badford protein assay. Equal amount of protein from each treatment was subjected to IL-1ßELISA based on manufacturer’s instructions. Compared to no smoke control, in 5% and 10% CSE treated CSCs, IL-1ßsecretion were significantly upregulated within 15 minutes (p < 0.05). Based on the results it can be concluded that acute cigarette smoke exposure promotes secretion of IL-1ß at 5% and10% concentrations.
INTRODUCTION
Cigarette smoking is a public health crisis and a leading cause of death around the world. Smoking leads to numerous illnesses including several Cardiovascular Diseases (CVD) [1]. Cigarette smoking related CVD include coronary artery disease / atherosclerosis [2-5], hypertension [6] and stroke [7]. Cigarette smoking-induced atherosclerosis occurs due to many mechanisms including modulation of prostaglandin levels [5], modification of the function of the endothelial cells, platelets, fibrinogen, and coagulation factors [8-10]. In addition, activation of endothelial inflammatory response, Il-8 [11], IL-6 [4] and IL-1ß production [12] leading to downstream signaling such as activation of Cyclooxygenase 2 (COX 2) enzyme [11] play a role in cigarette smoke-induced atherosclerosis. Cigarette Smoke (CS) caused attenuation of mitochondrial function both in-vitro (H9C2 heart cells) [13] and in-vivo (wild type FVB mice) [14]. In addition, CS induced endothelial cell death [15] and modulation of cardiac contractility [14] have been reported, all of these will lead to CS induced -CVD.
Although abundant literature is available linking CS exposure with CVD, there is limited literature on CS-induced changes in Cardiac Stem Cells (CSCs). Our focus is to investigate CS-induced modulations of Cardiac Stem Cell (CSC) function and responsible mechanisms. CS-induced adverse functional effects of c-kit+ Cardiac Stem Cells (CSCs) were reported for the first time by our group [16]. Cardiac Stem Cell Therapy (CSCT) is currently being used in clinical trials [17]. Adverse changes of CSCs caused by CS might lead to decreased potential of resident CSCs to respond to cardiac injury or to be used in CSCT. Therefore, studying CS-induced modulations of CSC functions and responsible mechanisms are of uttermost importance.
Our previous study reported that exposure to Cigarette Smoke Extract (CSE) caused adverse functional effects in CSCs. Those CSE- induced CSC modulations included increased apoptosis, decreased proliferation, increased cytotoxicity and gap junctional permeability [16]. The current study investigated whether CSCs produce interleukin 1 beta (IL-1ß) in response to exposure to CSE, with the view of understanding the role of interleukins as one of the causative agents in CSE-induced modulation of CSC signaling and functional effects.
IL-1ß and IL-18 are inflammatory cytokines that mature in inflammasomes, which are usually not present in healthy cells but are assembled upon infection, cellular stress, cell and tissue injury [18]. IL-1ß is one of the key mediators of inflammation, which is necessary to elicit host response to pathogens [19]. Upon stimulation by an insult, IL-1ß is secreted continuously from inflammasomes and the secretion depends on the stimulus strength and the extracellular IL-1 beta requirement [19]. IL-1ß plays a role in progression of cardiovascular disease. Increased secretion of Il-1ß was linked to the progression of many cardiovascular diseases including heart failure [20], ventricular arrhythmias due to QT interval prolongation [21] and coronary artery disease / atherosclerosis [22]. Our laboratory focuses on studying cigarette smoke induced cardiovascular effects and the focus of this project is to investigate whether cigarette smoke induces IL-1ß production in cardiac stem cells.
Tobacco induced production of inflammatory cytokines was observed in both in-vitro and in-vivo studies. Cigarette smoke induced inflammation was seen in several different cell types leading to many different diseases. Upregulation of IL-6 and IL-1ß due to cigarette smoke exposure have been reported in human gingival cells [23]. Increased interleukin 8 production due to cigarette smoke exposure was seen in dendritic cells [24] and endothelial cells [25]. Mice exposed to cigarette smoke as well as waterpipe tobacco smoke demonstrated elevated levels of pro-inflammatory markers such as TNF-α and IL-6 in the lung alveoli [26].
In a cardiac transplant study, it has been reported that pre-exposure of donor and recipient rats to cigarette smoke caused allograft rejection due to inflammation/interleukin production [27]. However to date the cigarette smoke-induced production of interleukin 1ß has not been studied in c-kit+ CSCs. The role of these interleukins in cigarette smoke-induced modulations of cardiac stem cell functions remains elusive. The current study tested the hypothesis that exposure to Cigarette Smoke Extract (CSE) will cause increased production of IL-1ß in CSCs.
MATERIALS AND METHODS
Materials
Access to c-kit+ CSCs: These CSCs of rat origin were gifted to the Institute of Molecular Cardiology, University of Louisville by Dr. Anversa, Birmingham Woman’s hospital, NY. Dr. Greg Rokosh, Institute of Molecular Cardiology, University of Louisville has generously provided this passage2 c-kit+ CSCs to our group. c-kit+ CSCs are susceptible to differentiate if they grow continuously [32]. In order to prevent differentiation, these CSCs were cultured only to a maximum of 15 passages, and used in the experiments outlined in this manuscript.
Cell culture media and other supplies: Ham’s F-12 media was purchased from Invitrogen / Thermo–Fisher scientific (Carlsbad, CA). Fetal bovine serum and horse serum were purchased from Atlanta Biological (Norcross, GA) and Invitrogen (Carlsbad, CA) respectively. Cell growth factors, namely basic fibroblast growth factor (bFGF) and human erythropoietin, were purchased from Pepro Tech (Rocky Hill, NJ) and Chemicon International (Billerica, MA) respectively. Bradford protein Assay reagents were purchased from Amresco (Solon, OH). ELISA kits were purchased from Boster Biological (Pleasanton, CA). L-glutathione was purchased from Sigma Aldrich (St. Louis, MO). Cell culture supplies were purchased from Midwest Scientific (Valley Park, MO) and VWR (Radnor, PA) companies. Cigarette smoke preparation apparatus were provided by Dr. Gregg Rokosh, University of Louisville.
Cigarette Smoke Extract (CSE) preparation: CSE were created by combusting camel filter free cigarettes and dissolving the smoke / puff in cell culture media based on a published method [16,34]. In brief, 10 ml of serum free cell culture media was drawn to a 60 ml syringe device, which then was attached to an adapter / stopcock that can hold a cigarette. Different volumes of smoke can be drawn by this method (Figure 1). Combustion of 1 whole cigarette (100%) yields about 300 ml puff [35]. Based on that, exposure concentrations of CSE were calculated for the current study. 2% of a cigarette = 6 ml puff / smoke, 5% of a cigarette = 15 ml of puff / smoke, and 10% of a cigarette = 30 ml of puff / smoke. Once the smoke was drawn to 10 ml serum free media, it was mixed with the media for 1-2 minutes. Then smoked media were filtered using 0.2 micron filter and kept in closed conical tubes. The purpose of filtration is twofold; to sterilize the smoked media as well as to get rid of any insoluble particles. For the no-smoke negative control, 10 ml of serum and growth factor free medium, which is subjected to the same filtration method, was used. For each experiment, CSE were made fresh and used within 5-10 minutes after the preparation.
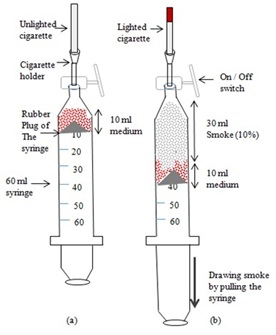
(b) Smoking apparatus showing extraction of 30 ml CSE.
Cardiac stem cell culture and CSE treatments: Purified CSCs from rat origin that express the c-kit marker [32] have been used. To preserve stem cell growth characteristics without differentiation, CSCs were cultured only up to passage 15 and utilized in the experiments. CSCs were cultured using Ham’s F-12 growth media (Invitrogen, CA) supplemented with 10% fetal bovine Serum (Atlanta Biological, GA), 5% horse serum (Invitrogen, CA), 0.2 mM L-Glutathione (Sigma Aldrich, MO), 10 ng / ml human bFGF (Pepro Tech, NJ), 0.005 U / ml human erythropoietin (Chemicon International, MA) at 37ºC with 5% CO2, 95% air, and above 90% humidity.
Equal amount of CSCs (40,000) were grown in 100 mm cell culture dishes for two days. Cell culture growth media was then removed, and cells were washed with Phosphate Buffered Saline (PBS) and transferred to the serum and growth factor (bFGF and erythropoietin) free HAM’s F-12 medium. Following overnight serum starvation, CSCs were exposed to either no-smoke control (0% CSE, just serum and growth factor - free HAM’s F12 medium) or varying concentrations of CSE (2%, 5%, and 10% dissolved in serum and growth factor-free HAM’s F12 medium) in the cell culture incubator. Depending on the experiment, CSCs were exposed to these treatments for either short term (15 minutes and 1hr) or 24 hours. In order to assure reproducibility, each experiment was performed 3-5 times. CSE concentrations selected for this study were clinically relevant [36] and similar to prior published reports [34,37]. After CSE exposure, the cell supernatants (media overlaying cells) were removed, and kept in - 80°C for IL-1ß detection experiments.
Bradford protein assay: After CSE exposure, protein amount from supernatants from each treatment group (no-smoke control, 2% CSE, 5% CSE, 10% CSE from each CSE exposure time point) were quantified using Bradford assay (Amresco, Solon, OH) based on manufacturer’s protocol.
IL-1ß Enzyme Linked Immunosorbent Assay (ELISA): IL-1ß secretion from CSCs due to CSE exposure was detected using the IL-1ß sandwich ELISA method. Equal amount of protein from different treatments were subjected to IL-1ß ELISA assay according to manufacturer’s instructions (Boster Biologicals, Pleasanton, CA). In brief, IL-1ß standards ranging from 0 pg /ml - 250 pg /ml were made from the lyophilized recombinant rat IL-1ß 10 ng standard using serial dilution. Proper blanks (e.g., some wells without any cell supernatant, just standard diluent buffer; some wells without Il-1 beta antibody) were used. Equal amount of protein from IL-1ß standards, no smoke negative control and test samples (CSE treated cell supernatants) were incubated in a 96 well plate pre-coated with the rat IL-1ß primary antibody. Then the biotinylated anti rat-IL-1ß antibody was added to the wells. Target protein (IL-1ß) from standards and treatment groups were captured/sandwiched by both antibodies. After necessary washes in 0.01 M Phosphate Buffered Saline (PBS) to get rid of non- specific binding, the plate was incubated with the Avidin-Biotin-peroxidase Complex (ABC). Following necessary washes in PBS, substrate solution for the peroxidase enzyme (color developing agent) was added and incubated until the color was developed. The reaction was then stopped using the stop solution and the colorimetric detection of IL-1ß was performed using a spectrophotometric plate reader.
Statistical analysis: In order to assure reproducibility each experiment was conducted 3 - 5 times. Statistical analysis was performed using sigma plot software. One-way Analysis of Variance (ANOVA) followed by Holm-Sidak method and Dunn’s method was performed.
P < 0.05 was considered significant.
RESULTS
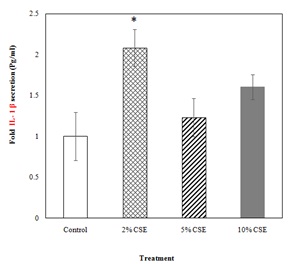
Aqueous extracts of cigarette smoke were prepared using the smoking apparatus, a 60 ml syringe attached to an adapter to hold the cigarette (Figure 1). CSCs were treated with these freshly prepared CSE for different time periods. Acute (either 15 minutes or 1 hour) CSE exposure to CSCs caused an increase in IL-1ß secretion in a dose dependent manner. At 15 minute CSE exposure, IL-1ß production of CSCs over no smoke control was significant at 5% and 10% CSE concentrations (Figure 2). Following 1 hr CSE exposure, CSE caused significant increase in IL-1ß secretion at 10% CSE concentrations (Figure 2). In addition, at 10% CSE treatment, IL-1ß secretion after 1 hour exposure was significantly higher than that of 15 min exposure. Long term (24 hour) exposure of CSCs to CSE showed significant increase in IL-1ß production only at 2% concentration (Figure 3).
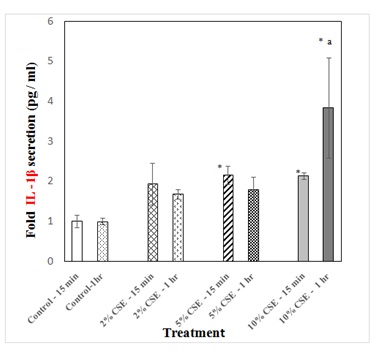
Figure 2: Detection of IL-1β production due to acute exposure to CSE. c-kit+ CSCs were treated with either the no smoke control or three different concentrations (2%, 5% and 10%) of CSE dissolved in 10 ml of serum free HAM’s F-12 media for 15 minutes and 1 hr. After CSE treatment, equal amount of protein from treatment groups (supernatants) were subjected to IL-1β ELISA. Secreted IL-1β was quantified, and the amount present in the no smoke control was taken as 1 and CSE-induced production of IL-1β relative to the no smoke control was plotted against the CSE concentration.
DISCUSSION
Tobacco smoke is an intricate mixture that contains thousands of chemicals [38], many of which are water soluble and present in aqueous extracts. A study that analyzed the water-soluble portion of cigarette smoke utilizing IR, mass and NMR spectra reveled that there were close to 500 water-soluble components in cigarette smoke [39]. These include groups such as acids, lactones, esters, amides, imides, aldehydes, ketones, alcohols, imidazole and pyridine derivatives [39]. Aqueous extracts of cigarette smoke contain chemicals including tar, which contains a mixture of semiquinones, hydroquinones and quinones that can produce free radicals, which were detected by Electron Paramagnetic Resonance (EPR) spectroscopy and Gas Chromatography/Mass Spectrometry (GC/MS) [40]. Analysis of the aqueous extraction from the tobacco that were used to fill cigarettes by Electrospray Ionization-Ion Trap Mass Spectrometry (ESI-ITMS) revealed that the tobacco extract contained quinic acid, citric acid and malic acid [41].
It was difficult to determine the exact chemical components of the CSE in our aqueous preparation since there were so many of them. However, it is very likely that all water soluble contents in particulate matter and gas phase must be dissolved in the culture medium. Our aqueous extract of cigarette smoke should contain all the water soluble components of cigarette smoke, including cotinine, hydralazine, heavy metals, anthracenes, tobacco specific nitrosamines, alkaloid, ammonia, dioxins, nicotine and pyrenes including benzo (a) pyrene; many of which are known toxicants and carcinogens [38,42].
Although CSE induced-cytotoxicity of many cells including cardio myoblasts [43] and endothelial cells [44] have been reported, our group was the first ones to report the CSE-induced malfunction of c-kit+CSCs [16]. We have reported that CSE exposure has led to attenuated CSC proliferation and migration, increased CSC apoptosis, cytotoxicity and caused damage in CSC membrane resulting in dysfunctional CSCs [16].
In 2015, based on an in-vitro differentiation assay, palpant and colleagues reported that CSE (both tobacco cigarettes and e-cigarettes) caused malfunction of human embryonic stem cells leading to malformed hearts [45]. This study also investigated CSE induced in-vivo effects using zebra fish. They have reported that CSE caused severe heart malformation and reduced heart function of zebra fish, and the toxic effects were more severe in tobacco cigarettes compared to e cigarettes [45]. CSE also caused reduced heart rate and decreased expression of cardiac transcription factors and channels including GATA4 and L-type calcium channels [45].
The focus of our laboratory is to investigate the CSE induced damage to c-kit+ CSCs and to unveil the possible mechanism. Properly functioning CSCs are required for cardiac stem cell therapy. Cardiac stem cell therapy is one of the promising therapeutic options available to treat a damaged heart following Myocardial Infarction (MI) [17]. Stem cell therapy using c-kit+ cardiac stem cells has been conducted in post MI patients and proven to increase cardiac function of the stem cell therapy recipients [17].
Since we have previously reported CSE induced functional changes of CSCs, the current study was focused on investigating the possibility of CSE induced modulation of IL-1ß secretion as a possible mechanism for CSE induced changes in CSCs. IL-1ß is a potent pro-inflammatory cytokine and it is upregulated in the infarcted myocardium due to MI- induced activation of cardiomyocytes and interstitial cell inflammasomes [46]. In addition to cytokines, ischemic damage due to MI leads to recruitment of huge amount of leukocytes. A recent study utilizing Apolipoprotein -E (ApoE) knock out atherosclerotic mice, suggests that IL-1ß was involved in hematopoietic stem cell proliferation leading to large amount of leukocyte production. Targeting IL-1ß provides a way to decrease inflammatory damage after MI [47]. There were some reports discussing CSE induced IL-1ß production in other cell models [48,49]. However, the current study is the first report discussing CSE induced IL-1ß release in c-kit+ CSCs. Here we tested the hypothesis that CSE caused an increase in IL-1ß secretion in CSCs.
Based on the results of the current study, it can be concluded that cigarette smoke (5% and 10%) promotes production of IL-1ß in c-kit+ CSCs within a short period of time (Figure 2). It is a novel finding and we are the first ones to report CSE induced IL-1ß recruitment in c-kit+ CSCs. IL-1ß is a secretory cytokine and 2% CSE may not be sufficient to cause significant increase in IL-1ß in CSCs for exposures of 15min or 1 hr. However, when exposed to CSCs for 24 hours, it increases Il-1ß production significantly (Figure 3).
Following 1 hr exposure of CSE, there is a dose dependent increase in IL-1ß release from CSCs, which is significant at 10% CSE concentration. In agreement with our results, the CS-induced production of IL-1ß has been reported in macrophages [48] and blood mononuclear cells [49]. CS induced IL-8 has also been reported in Human Umbilical Vein Endothelial Cells (HUVEC) [11,25].
Based on an inflammatory transcriptome profiling study using CSE and Human Monocytes (THP-1 cells), it has been reported that numerous genes, which are involved in innate immune system, were activated by 10% CSE exposure. Following 8 hr exposure to 10% CSE, pro-inflammatory cytokines such as TNF and IL-12 genes were upregulated [50]. Following 24 hours of exposure to 10% CSE, the gene profile has somewhat changed and genes involved in adaptive immune system were activated, no longer showing an increase in pro inflammatory IL- 12. Their data was in agreement with the recent hypothesis of “monocytes and macrophages change their profile from a pro-inflammatory phenotype (caused by innate immunity) to a tissue remodeling phenotype (caused by adaptive immunity) when in diseased tissue” [51]. Both innate and adaptive immunity play a crucial role in cardiac injury and repair [52]. Cardiac injury induced gene profile switch from pro-inflammatory genes to tissue repair genes occurred with the long term exposure of 10% CS [50].
This is in agreement with our results. In our study, 24 hr exposure of both 10% and 5% CSE did not cause significant increase in IL-1ß production in CSCs. The reason for non-significant IL-1ß production at 24 hr exposure to higher CSE concentrations could be due to more dead cells causing the switch in pro-inflammatory genes to tissue repair genes, as seen by wright and colleagues in their experiments [50]. Our group previously reported the increased apoptotic death of the same cell line at 5% and 10% CSE concentrations [16].
Any of the other pro-inflammatory genes in interleukin family (Il-1ß, IL-6, or IL-8) were not upregulated or downregulated by CSE treatment ininflammatory transcriptome profiling study [50]. According to another report, 6 hr and 24 hr. CSE exposure to cultured carotid arteries resulted in significant increase in IL-1ß and IL-6 mRNA [12]. Since we have observed significant increase in CSE induced Il-1ß production in CSCs just after 15 minutes, it is very unlikely that this Il-1ß production is a result of transcription, because transcription takes long time (hours). Therefore, ELISA was chosen over RT-PCR, so we can detect CSE-induced modulation of IL-1ß protein level.
IL-1ß plays a major role in inflammatory damage after myocardial infarction and it is upregulated in the infarcted myocardium [46]. Since c-kit+ CSCs are currently being used in CSCT trials it is important to investigate the ways to improve function of c-kit+ CSCs. The current study will provide mechanistic insight for CSE induced malfunction of c-kit+ CSCs reported earlier [16]. CSE induced IL-1ß production is just one of the possible mechanisms for CSE induced CSC malfunction. CSE induced modulation of other cytokines, cell signaling, and oxidative stress may serve as other possible mechanisms. Those mechanisms and the effects of IL-1ß inhibition on CSC functions will be investigated in the near future.
ACKNOWLEDGEMENT
Funding was provided by the Sullivan University Faculty Development Grant (RG_1_PS_2012_03).
Special thanks go to Dr. Anversa and Dr. Rokosh for providing c-kit+ CSCs. Dr. Rokosh also provided the smoking apparatus and also served as a collaborator of the funded grant. We would like to thank Dr. Zhao, Sullivan University College of Pharmacy for serving as a collaborator for the funded grant. We acknowledge Dr. Houston Acha for his help with preparation of chemicals and Thimira Sumanasekera for editing the manuscript.
REFERENCES
- Landini L, Leone A (2011) Smoking and hypertension: effects on clinical, biochemical and pathological variables due to isolated or combined action on cardiovascular system. Curr Pharm Des 17: 2987-3001.
- Song W, Wang W, Dou LY, Wang Y, Xu Y, et al. (2015) The implication of cigarette smoking and cessation on macrophage cholesterol efflux in coronary artery disease patients. J Lipid Res 56: 682-691.
- Lee YA, Kang SG, Song SW, Rho JS, Kim EK (2015) Association between metabolic syndrome, smoking status and coronary artery calcification. PLoS One 10: 0122430.
- Ambrose JA, Barua RS (2004) The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol 43: 1731-1737.
- Amadio P, Baldassarre D, Tarantino E, Zacchi E, Gianellini S, et al. (2015) Production of prostaglandin E2 induced by cigarette smoke modulates tissue factor expression and activity in endothelial cells. FASEB J 29: 4001-4010.
- Talukder MA, Johnson WM, Varadharaj S, Lian J, Kearns PN, et al. (2011) Chronic cigarette smoking causes hypertension, increased oxidative stress, impaired NO bioavailability, endothelial dysfunction and cardiac remodeling in mice. Am J Physiol Heart Circ Physiol 300: 388-396.
- Shah RS, Cole JW (2010) Smoking and stroke: the more you smoke the more you stroke. Expert Rev Cardiovasc Ther 8: 917-932.
- Barua RS, Ambrose JA (2013) Mechanisms of coronary thrombosis in cigarette smoke exposure. Arterioscler Thromb Vasc Biol 33: 1460-1467.
- Barua RS, Sy F, Srikanth S, Huang G, Javed U, et al. (2010) Acute cigarette smoke exposure reduces clot lysis--association between altered fibrin architecture and the response to t-PA. Thromb Res 126: 426-430.
- Barua RS, Sy F, Srikanth S, Huang G, Javed U, et al. (2010) Effects of cigarette smoke exposure on clot dynamics and fibrin structure: an ex vivo investigation. Arterioscler Thromb Vasc Biol 30: 75-79.
- Barua RS, Sharma M, Dileepan KN (2015) Cigarette smoke amplifies inflammatory response and atherosclerosis progression through activation of the H1R-TLR2/4-COX2 Axis. Front Immunol 6: 572.
- Orosz Z, Csiszar A, Labinskyy N, Smith K, Kaminski PM, et al. (2007) Cigarette smoke-induced proinflammatory alterations in the endothelial phenotype: role of NAD(P)H oxidase activation. Am J Physiol Heart Circ Physiol 292: 130-139.
- Tippetts TS, Winden DR, Swensen AC, Nelson MB, Thatcher MO, et al. (2014) Cigarette smoke increases cardiomyocyte ceramide accumulation and inhibits mitochondrial respiration. BMC Cardiovasc Disord 14: 165.
- Hu N, Han X, Lane EK, Gao F, Zhang Y, et al. (2013) Cardiac-specific over expression of metallothionein rescues against cigarette smoking exposure-induced myocardial contractile and mitochondrial damage. PLoS One 8: 57151.
- Messner B, Frotschnig S, Steinacher-Nigisch A, Winter B, Eichmair E, et al. (2012) Apoptosis and necrosis: two different outcomes of cigarette smoke condensate-induced endothelial cell death. Cell Death Dis 3: 424.
- Sumanasekera WK, Tran DM, Sumanasekera TU, Le N, Dao HT, et al. (2014) Cigarette smoke adversely affects functions and cell membrane integrity in c-kit+ cardiac stem cells. Cell Biol Toxicol 30: 113-125.
- Bolli R, Chugh AR, D’Amario D, Loughran JH, Stoddard MF, et al. (2011) Cardiac Stem Cells in Patients with Ischaemic Cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet 378: 1847-1857.
- Horstmann JP, Marzi I, Relja B (2016) Adrenergic stimulation alters the expression of inflammasome components and interleukins in primary human monocytes. Exp Ther Med 11: 297-302.
- Lopez-Castejon G, Brough D (2011) Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev 22: 189-195.
- Butts B, Gary RA, Dunbar SB, Butler J (2015) The Importance of NLRP3 inflammasome in heart failure. J Card Fail 21: 586-593.
- Sordillo PP, Sordillo DC, Helson L (2015) Review: the prolonged qt interval: role of pro-inflammatory cytokines, reactive oxygen species and the ceramide and sphingosine-1 phosphate pathways. In Vivo 29: 619-636.
- Enayati S, Seifirad S, Amiri P, Abolhalaj M, Mohammad-Amoli M (2015) Interleukin-1 beta, interferon-gamma and tumor necrosis factor-alpha gene expression in peripheral blood mononuclear cells of patients with coronary artery disease. ARYA Atheroscler 11: 267-274.
- Semlali A, Witoled C, Alanazi M, Rouabhia M, et al. (2012) Whole cigarette smoke increased the expression of TLRs, HBDs, and proinflammory cytokines by human gingival epithelial cells through different signaling pathways. PLoS One 7: 52614.
- Mortaz E, Lazar Z, Koenderman L, Kraneveld AD, Nijkamp FP, et al. (2009) Cigarette smoke attenuates the production of cytokines by human plasmacytoid dendritic cells and enhances the release of IL-8 in response to TLR-9 stimulation. Respir Res 10: 47.
- Wang H, Ye Y, Zhu M, Cho C (2009) Increased interleukin-8 expression by cigarette smoke extract in endothelial cells. Environ Toxicol Pharmacol 9: 19-23.
- Khabour OF, Alzoubi KH, Bani-Ahmad M, Dodin A, Eissenberg T, et al. (2012) Acute exposure to waterpipe tobacco smoke induces changes in the oxidative and inflammatory markers in mouse lung. Inhal Toxicol 24: 667-675.
- Khanna AK, Xu J, Uber PA, Burke AP, Baquet C, et al. (2009) Tobacco smoke exposure in either the donor or recipient before transplantation accelerates cardiac allograft rejection, vascular inflammation and graft loss. Circulation 120: 1814-1821.
- Dawn B, Zuba-Surma EK, Abdel-Latif A, Tiwari S, Bolli R (2005) Cardiac stem cell therapy for myocardial regeneration. A clinical perspective. Minerva Cardioangiol 53: 549-564.
- Dawn B, Bolli R (2005) Cardiac progenitor cells: the revolution continues. Circ Res 97: 1080-1082.
- Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, et al. (2003) Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114: 763-776.
- Barile L, Chimenti I, Gaetani R, Forte E, Miraldi F, et al. (2007) Cardiac stem cells: isolation, expansion and experimental use for myocardial regeneration. Nat Clin Pract Cardiovasc Med 1: 9-14.
- Tang XL, Rokosh G, Sanganalmath SK, Yuan F, Sato H, et al. (2010) Intracoronary administration of cardiac progenitor cells alleviates left ventricular dysfunction in rats with a 30-day-old infarction. Circulation 121: 293-305.
- Dawn B, Stein AB, Urbanek K, Rota M, Whang B, et al. (2005) Cardiac stem cells delivered intravascularly traverse the vessel barrier, regenerate infarcted myocardium, and improve cardiac function. Proc Natl Acad Sci U S A 102: 3766-3771.
- Yoon CH, Park H-J, Cho Y-W, Kim E-J, Lee JD, et al. (2011) Cigarette smoke extract-induced reduction in migration and contraction in normal human bronchial smooth muscle cells. Korean J Physiol Pharmacol 15: 397-403.
- Liu X, Conner H, Kobayashi T, Kim H, Wen F, et al. (2005) Cigarette smoke extract induces DNA damage but not apoptosis in human bronchial epithelial cells. Am J Respir Cell Mol Biol 33: 121-129.
- Yamada S, Zhang XQ, Kadono T, Matsuoka N, Rollins D, et al. (2009) Direct toxic effects of aqueous extract of cigarette smoke on cardiac myocytes at clinically relevant concentrations. Toxicol Appl Pharmacol 236: 71-77.
- Liu X, Togo S, Al-Mugotir M, Kim H, Fang Q, et al. (2008) NF-kappaB mediates the survival of human bronchial epithelial cells exposed to cigarette smoke extract. Respir Res 9: 66.
- Talhout R, Schulz T, Florek E, van Benthem J, Wester P, et al. (2011) Hazardous compounds in tobacco smoke. Int J Environ Res Public Health 8: 613-628.
- Schumacher JN, Green CR, Best FW, Newell MP (1977) Smoke composition. An extensive investigation of the water-soluble portion of cigarette smoke. J Agric Food Chem 25: 310-320.
- Pryor WA, Stone K, Zang LY, Bermúdez E (1998) Fractionation of aqueous cigarette tar extracts: fractions that contain the tar radical cause DNA damage. Chem Res Toxicol 11: 441-448.
- Ng LK, Lafontaine P, Vanier M (2004) Characterization of cigarette tobacco by direct Electrospray Ionization-Ion Trap Mass Spectrometry (ESI-ITMS) analysis of the aqueous extract--a novel and simple approach. J Agric Food Chem 52: 7251-7257.
- Raveendran M, Senthil D, Utama B, Shen Y, Wang J, et al. (2005) Effect of water-soluble fraction of cigarette smoke on human aortic endothelial cells--a proteomic approach. Cell Biol Toxicol 21: 27-40.
- Farsalinos KE, Romagna G, Allifranchini E, Ripamonti E, Bocchietto E, et al. (2013) Comparison of the cytotoxic potential of cigarette smoke and electronic cigarette vapour extract on cultured myocardial cells. Int J Environ Res Public Health 10: 5146-5162.
- Green LA, Petrusca D, Rajashekhar G, Gianaris T, Schweitzer KS, et al. (2012) Cigarette smoke-induced CXCR3 receptor up-regulation mediates endothelial apoptosis. Am J Respir Cell Mol Biol 47: 807-814.
- Palpant NJ, Hofsteen P, Pabon L, Reinecke H, Murry CE (2015) Cardiac development in zebra fish and human embryonic stem cells is inhibited by exposure to tobacco cigarettes and e-cigarettes. PLoS One 10: 0126259.
- Frangogiannis NG (2015) Interleukin-1 in cardiac injury, repair and remodeling: pathophysiologic and translational concepts. Discoveries (Craiova) 3.
- Sager HB, Heidt T, Hulsmans M, Dutta P, Courties G, et al. (2015) Targeting Interleukin-1β Reduces Leukocyte Production After Acute Myocardial Infarction. Circulation 132: 1880-1890.
- Lau PP, Li L, Merched AJ, Zhang AL, Ko KW, et al. (2006) Nicotine induces proinflammatory responses in macrophages and the aorta leading to acceleration of atherosclerosis in low-density lipoprotein receptor (-/-) mice. Arterioscler Thromb Vasc Biol 26: 143-149.
- Ryder MI, Saghizadeh M, Ding Y, Nguyen N, Soskolne A, et al. (2002) Effects of tobacco smoke on the secretion of interleukin-1beta, tumor necrosis factor-alpha and transforming growth factor-beta from peripheral blood mononuclear cells. Oral Microbiol Immunol 17: 331-336.
- Wright WR, Parzych K, Crawford D, Mein C, Mitchell JA, et al. (2012) Inflammatory transcriptome profiling of human monocytes exposed acutely to cigarette smoke. PLoS One 7: 30120.
- Shaykhiev R, Krause A, Salit J, Strulovici-Barel Y, Harvey BG, et al. (2009) Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol 183: 2867-2883.
- Epelman S, Liu PP, Mann DL (2015) Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol 15: 117-129.
Citation: Sumanasekera W, Waingeh B (2016) Does Cigarette Smoke Cause Interleukin 1 - Beta (Il-1ß) Production in Cardiac Stem Cells? J Cell Biol Cell Metab 3: 012.
Copyright: © 2016 Wasana Sumanasekera, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.