Mutation Identification for Epilepsy in the US Hispanic Population Using Whole-Exome-Sequencing
*Corresponding Author(s):
Chun XuDepartment Of Pediatrics, Paul L. Foster School Of Medicine, Texas Tech University Health Sciences Center, Texas, United States
Tel:+1 9152154196,
Email:chun.xu@ttuhsc.edu
Abstract
Purpose: Epilepsy, characterized by recurrent unprovoked seizures, is a common neurological disorder related to a wide variety of genetic, developmental and acquired brain conditions. Genetically determined epilepsies are associated with a multiplicity of potential genetic variants identified in predominately Caucasian populations. There has been limited research in the US Hispanics/Latino populations. Whole Exome Sequencing (WES), as a rapid and reliable means, has identified causative mutations for a number of diseases. Therefore, we applied WES to identify pathogenic mutations for epilepsy in the Latino population.
Methods: Among 14 symptomatic subjects recruited from the Departments of Psychiatry and Neurology at the Texas Tech University Health Sciences Center in El Paso, Texas, 11 met the diagnosis criteria for epilepsy. WES was performed by the Illumina Nextera Rapid Capture Exome Enrichment kits, then bioinformatics predictions were conducted using the SVS program by accessing data from dbNSFP, which provides scores from multiple functional prediction programs and two conservation scores.
Results: A total of 47,392 variants in exons or at splice-site boundaries were identified. After filtering, 18 non-synonymous rare variants were identified in 8 out of 11 patients. To identify high impact genes and biological pathways, further bioinformatics analysis, “hotspot” analysis and literature-based searches were conducted. The CREBBP gene and pathways, such as “cell cycle” and “Notch signaling” were suggested to be important in the pathophysiology of epilepsy.
Conclusion: This WES led to the discovery of additional epilepsy genes in the US Latino population; however, future validation and segregation analysis using a family design are needed to confirm the current findings.
Keywords
INTRODUCTION
Epilepsy, characterized by recurrent unprovoked seizures, is the most common neurologic disorder after headache, with a prevalence of 5 to 10 in 1000 persons and an incidence of 50 to 120 in 100,000 per year in the United States [1]. Abundant evidence of a genetic contribution to epilepsy in humans derives from family and twin studies shows concordance rates for epilepsy of 50 to 60% in monozygotic twins and 15% in dizygotic twins [2]. Given its affecting a variety of mental and physical functions, this disease extracts a marked toll in terms of morbidity and economic burden and imposes tremendous burden on patients and the health system in general.
Previous studies of epilepsy have suggested the notion that genetics play a major role in the disease, largely by identifying channels and neurotransmitters important in epileptogenesis. However, the mechanism of the disease-associated variants (single nucleotide polymorphisms, SNPs and/or copy number variations) in these channels and neurotransmitters is poorly understood. With the tremendous advance of technological development, the results of Genome-Wide Association (GWA) and candidate gene studies have suggested a few potential risk variants for epilepsy and encompass two broad categories reported in predominately Caucasian populations: 1) the genes/loci discovered in association with primary epilepsy syndromes; 2) the genes suggested in association with disorders of brain development that are associated with epilepsy [3]. However progress in extending beyond identification of disease-causing mutations and clarify disease pathophysiology mechanisms has been slow. In addition, disease susceptibility alleles individually contribute only modestly to the overall disease risk and very limited markers discovered show meaningful predictive genetic power for the disease based on conventional gene-discovery strategies (e.g., GWA, candidate gene studies). To date, 14 GWA studies, 38 meta-analyses, 339 genes in epilepsy have been reported based on Huge Navigator. In addition, a numbers of genes/loci that contribute to the genetic basis of epilepsy collectively account for only a small fraction of the observed heritability of this disease. Furthermore, little is known about the extent to which rare alleles contribute to the heritability of epilepsy and rare and potentially deleterious variants in protein coding regions may not be detected by GWA studies. Recent studies have focused on genetic factors beyond the channelopathies.
Recently, progress towards a full resolution of the genetic basis of human complex diseases is being substantially aided by development of next generation sequencing technologies, including whole exome sequencing. Using whole genome sequencing technology and parent-offspring design, the researchers at the University of Arizona discovered a de novo heterozygous missense mutation (c.5302A>G [p.Asn1768Asp]) in the voltage-gated sodium-channel gene SCN8A in a proband, a 15-year-old female with a severe epileptic encephalopathy consisting of early-onset seizures, features of autism, intellectual disability, ataxia and sudden unexplained death in epilepsy [4].
In order to better understand the etiology of epilepsy, a recent study in Maryland evaluated 18 genes highly suspected to be associated with major forms of primary and syndromic epilepsy in study participants. Of the 1,600 participants, 261 (16%) were carriers of the anomalous genes, while patients with infantile onset epilepsy had an even higher positive diagnostic rate of about 20% [5].
It was presumed from these results that 20% of all cases had a Mendelian origin. However, the question remains as to whether or not more genetic aberrancies besides the initially suspected 18 could be responsible for symptoms of epilepsy. Since only 20% of epilepsy cases can be explained with genetic research, new methods of sequencing should be employed in order to identify additional genetic causes of disease.
By evaluating the portions of DNA encoding for proteins in the human genome, a study participant can be screened for mutations much faster and cost-effectively than conventional genome sequencing (<$600 per sample vs $3,000-6,000 per sample conventionally) [6]. Coupled with advanced sequencing analysis software techniques, rare mutations can be located within whole exomes and shed light on proteins and therefore disrupted metabolic pathways presumed to be responsible for the disease. Whole exome sequencing, which generates sequence data from hundreds of millions of short DNA fragments, promises to speed up discovery of the genetic causes of disease in both the basic research and the clinical setting [7]. Increased speed and decreased costs of whole exome sequencing are leading to identification of novel mutations for monogenic and polygenic diseases [8-14] including epileptic encephalopathies [12] (for review, see [15]), application in clinical practice and improved medical care [16].
Importantly, limited research has been done in the US Latino population to date and suggests that general health information and knowledge of etiology and genetic bases of epilepsy among the Latinos have been insufficient. A previous study reported that 400,000 Latino-Americans are among the nation’s 2.7 million affected with epilepsy (15%). Many of these individuals fear epilepsy because of traditional associations between seizures and death; a majority of surveyed Latinos were unwilling to disclose that a family member has the disorder (cited from epilepsy and the Latino community, health awareness, 2006, http://www.napsnet.com/pdf_archive/60/67222.pdf). Consequently, it is poorly understood if US Latino/Hispanics and Non-Hispanic Americans who have epilepsy have different risks and/or disease-causing mutations. Therefore, we focused on patients with epilepsy from the ~80% Latino (mostly Mexican heritage) El Paso-Las Cruces metropolitan population and uncover the genetic basis for the diseases.
In order to gain more insight into the exact genetic basis at the global level in epilepsy in the US Latino population, we applied whole exome sequencing as one of Next Generation Sequencing (NGS) technologies, together with newly developed statistical and bioinformatics tools in 14 adult patients with epilepsy. The objective of the study was to apply whole exome sequencing as unbiased means to detect genetic common and rare variants and to identify disruptive de novo mutation for epilepsy in the US Latino population.
MATERIALS AND METHODS
Subjects
Subjects were recruited from the outpatient clinic of the Department of Neurology at the Texas Tech University Health Sciences Center in El Paso, Texas. Detailed phenotypic information was collected at the time of enrollment and put into a database in a de-identified manner for the facilitation of genetics studies. Clinical information is updated periodically when the individual returns to the clinic for routine care and treatment. From the database information, we selected 14 related individuals with epilepsy from the US Latino population for the whole exome-sequencing study. After a careful review of the medical records, three of the initial 14 patients did not meet the diagnosis criteria for epilepsy.
Therefore, the total number of patients with epilepsy were 11 (3 males, 8 females) had met the following criteria and was selected for this study (Table 1): 1) clinical symptoms with documentation of EEG with epileptiform abnormities based on the criteria of the 1989 International League against Epilepsy syndrome definitions [17]. The subjects exhibited different severities of generalized seizures in addition to several other comorbidities; however in the study design, we made sure to consider patients with a past medical history of tonic-clonic (grand mal) seizures and majority was Latino ancestry; 2) neurodevelopmentally normal with no other identifiable/symptomatic explanation for their epilepsy which is consistent with current International League Against Epilepsy (ILAE) terminology for idiopathic generalized epilepsy; 3) generalized seizure spectrum including any combination of the following three seizure types: generalized tonic-clonic, myoclonic and absence. Diagnoses were assigned by the trained neurologists at TTUHSC. Age at onset of illness was defined as the first distinct episode meeting the criteria for epilepsy discussed above.
| Sex | Sub_ID | Age | Family history of seizures | Initial symptoms | Initial EEG | MRI |
| M | 2 | 33 | - | Seizures occur in clusters without aura and with hemifacial spasm | Seizure activity with myoclonus | Slightly larger R temporal horn with normal hippocampus |
| F | 3 | 71 | - | Partial without aura | Not documented | Cerebrocerebellar atrophy but no pathology |
| F | 4 | 43 | - | Unprovoked and abrupt loss of consciousness with collapse, Left side predominant stiffness and jerking and incontinence of urine. Sometimes can occur with fever | Abnormal: diffuse generalized theta activity, highly suggestive of post-ictal state. | Normal |
| M | 5 | 33 | (+) aunt with febrile seizures | History of seizures that recently reappeared after 10 year period, Myoclonic head jerks to right side, Asymmetric pupils R4mm L2mm, | Normal | Normal |
| F | 6 | 63 | (+) brother | Anticipation, auditory hallucination, does not fall or lose consciousness | Sporadic left frontotemporal sharp waves, multiple artifacts (less reliable findings) | Normal |
| M | 7 | 52 | - | Tonic-clonic seizures with urinary incontinence and tongue biting | Normal | Normal |
| F | 8 | 44 | - | Occasional tonoclonic activity with urinary incontinence and postictal lethargy and confusion | Abnormal. Paroxysmal activity consistent with primary epilepsy. | Normal |
| F | 9 | 56 | - | Patient was not hospitalized for seizure but wrist trauma. History of seizure is reported by patient. | Sporadic high amplitude R frontotemporal sharp waves, Intermittent irregular R anterior quadrant slowing | Right mesial temporal lobe sclerosis, incidental pituitary microadenoma, chronic ischemic white matter changes. |
| F | 11 | 45 | (+) multiple family members | Complex partial type with aura | Normal | Normal |
| F | 12 | 47 | - | Partial with aura | Not available for her EEG in the medical chart | Changes post right temporal craniectomy shows encephalomalacia without residual or recurrent signs of active nerocysticercosis, Small arachnoid cysts noted in right temporal fossa and Meckel’s cave bilaterally, Focal encephalomalacia within R superior temporal gyrus |
| F | 13 | 51 | - | Both type of seizures occur with menses | Mild generalized slowing | Normal |
“Hotspots”, define as at least two studies in several patients reported the cytogenetic locations were identified in epilepsy related phenotypes.
“+” means family member(s) has/have seizure related phenotypes; “-“ means family member(s) has/have no seizure related phenotype.
Whole exome sequencing
To identify potentially damaging rare variants, we first identified variants that were possibly functional (non-synonymous, frameshift, and splice site) and rare (minor allele frequency <1% in the 1000 genomes Project (http://www.1000genomes.org/). We then computationally analyzed Single Nucleotide Variants (SNVs) and insertions or deletions (indels) using algorithms that account for biochemical, evolutionary, and structural information [19] using Genome Browse-SVS (Version 8.2.1) (http://www.goldenhelix.com/GenomeBrowse/; Golden Helix, Bozeman, MT). Moreover, the observed variants were evaluated for their frequency in the general population using publicly available databases such as dbSNP (http://www.ncbi.nlm.nih.gov/snp), Exome Variant Server (EVS), Seattle, WA, (http://evs.gs. washington.edu/EVS/), National Heart, Lung and Blood Institute (NHLBI) Exome Sequencing Project (ESP), 6500 exomes, and 1000 Genomes project (http://www.1000genomes.org/data). Then we analyzed variants for their possible effects on protein function, which was performed within the SVS program by accessing data from dbNSFP, which provides scores from multiple functional prediction programs (SIFT, Polyphen2, Mutation Taster, Mutation Assessor and FATHMM) as well as two conservation scores (GERP++ and PhyloP) [20].
Pathway enrichment analysis
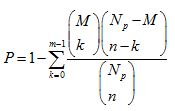
Where Np is the total number of genes in the whole genome, N is the number of epilepsy-associated genes, M is the number of genes annotated in the pathway and M is the number of epilepsy-associated genes in the pathway. If p value was < 0.05, the pathway was identified as a significantly enriched pathway.
A subnetwork of epilepsy-associated genes
RESULTS
From the initial import, the variants identified by whole exome sequencing not shared between study participants were removed, thus revealing 47,392 variants in common, within coding regions of DNA (exons) or near splice site boundaries. The resultant exon-enriched DNA libraries were sequenced using an Illumina MiSeq next generation sequencer to a median coverage of 126×.
Of these original 47,392 variants, four database probe tracks (SIFT, OMIM, NHLBIESP6500SI-V2 Exomes Variant Frequencies, and dbNSFPNS functional predictions 2.4., GHI) were used to remove common variant mutations that were unlikely to attribute to loss of function mutations or gain of aberrant function.
Data probe filtering yielded 18 non-synonymous rare variants within the study population in eight out of the 11 adult patients with epilepsy. All eight patients carried rare variants and these variants were confined to 12 different chromosomes and are displayed in table 2. There was a high quality of the current whole exome sequencing and an average read depth was 126×. Three (patient # 8, 11 and 12) out of 11 patients carried two and/or more mutations. Subject #12 carried three gene mutations, two (MFGE8 and CREBBP) of which are predicated damaging mutations by five functional prediction programs. Subject #8 carried seven gene mutations, three of which showed combined damage score > 3.5 (Table 2) for GPRC5D, SPG20 and NPC1 genes and two of which showed combined damage score >2.5 for genes of POTEH and PCSK1. The biological functions of those five genes include lipid metabolic process, cellular process, transport and regulations of transcription (Table 2).
| Gene Name | Entrez Gene ID | Putative Biological Function | patients # | Chr. Loc a. | Position (hg19) | Nucleotide Change | Protein Change | CFSb |
| CYP21A2 | 1589 | Metabolism | 3 | 6p21.33 e | chr6:32007967-SNVc | c.924G>T | p.Leu308Phe | 4.5 |
| MFGE8 | 4240 | nervous system development | 12 | 15q26.1 e | chr15:89442997-SNV | c.916G>T | p.Gly306Trp | 4.5 |
| NPC1 | 4864 | lipid metabolic process | 8 | 18q11.2 | chr18:21124402-SNV | c.2036G>A | p.Gly679Glu | 4.5 |
| COL9A2 | 1298 | cell-cell signaling | 11 | 1p34.2 | chr1:40780025-SNV | c.185C>T | p.Pro62Leu | 4 |
| CREBBP | 1387 | regulation of transcription | 12 | 16p13.3 e | chr16:3807959-SNV | c.3346G>A | p.Val1116Met | 3.5 |
| GPRC5D | 55507 | cellular process | 8 | 12p13.3 | chr12:13103023-SNV | c.296T>C | p.Leu99Pro | 3.5 |
| SPG20 | 23111 | Transport | 8 | 13q13.3 | chr13:36886587-SNV | c.1511G>T | c.1511G>T | 3.5 |
| HERC2 | 8924 | catabolic process, proteolysis | 11 | 15q13.1 | chr15:28474440-SNV | c.5173C>T | p.Arg1725Trp | 2.5 |
| MCM4 | 4173 | DNA replication, cell cycle | 13 | 8q11.21 | chr8:48882586-SNV | c.1403C>A | p.Pro468Gln | 2.5 |
| PCSK1 | 5122 | metabolic process | 8 | 5q15 | chr5:95759147-SNV | c.272C>T | p.Thr91Met | 2.5 |
| POTEH | 23784 | regulation of transcription | 8 | 22q11.1 | chr2:132021946-SNV | c.2918G>A | p.Gly973Asp | 2.5 |
| NLRP2 b | 55655 | cellular defense response | 8 | 19q13.42 e | chr19:55493942-SNV | c.876G>T | p.Glu292Asp | 1.5 |
| RASGRP1 | 10125 | cell communication | 11 | 15q14 | chr15:38808442-SNV | c.631G>A | p.Glu211Lys | 1.5 |
| ZNF276, FANCA | 92822 | regulation of transcription | 5 | 16q24.3 | chr16:89804312-SNV | c.1348C>A | p.Gln450Lys | 1.5 |
| EDC3 | 80153 | nucleic acid transport | 2 | 15q24.1 | chr15:74948339-SNV | c.555G>T | p.Lys185Asn | 1 |
| LRP2 | 4036 | metabolic process | 8 | 2q31.1 e | chr2:170030596-SNV | c.10847T>A | p.Phe3616Tyr | 1 |
| EVC2 | 132884 | alternative splicing, disease mutation | 6 | 4p16.2 | chr4:5578078-SNV | c.3161G>T | p.Gly1054Val | 0.5 |
| FLG | 2312 | Structural Molecule Activity | 12 | 1q21.3 | chr1:152280833-SNV | c.6529T>C | p.Ser2177Pro | 0 |
bCombined Function Score (CFS): the scores are based on five prediction function programs (SIFT, Polyphen2, Mutation Taster, Mutation Assessor, and FATHMM), e.g., 4.5 means damaging or disease causing from four programs, one of five shows probably damaging. For detail of prediction function from five programs, please see Supplementary table S1.
cSingle Nucleotide Variation (SNV).
e“hotspots”, define as at least two studies in several patients reported the cytogenetic locations were identified in epilepsy related phenotypes.
In addition, three out of the 11 patients did not carry any of the 18 mutations and they were subjects #4, 7 and 9. Clinically, there were no specific similarities or differences among the patients who harbored less pathogenic mutations as compared the rest of 8 patients who carried more pathogenic mutations. However, comorbidity of depression, anxiety or PTSD was less common in the patients (33%, #4, 7 and 9) with less pathogenic mutations as compared with patients (55%, the rest of 8 patients) who harbored more pathogenic mutations.
These 18 mutations were not found in publicly available databases, including 6503 individuals from the National Heart, Lung and Blood Institute Exome Sequencing Project, dbSNP (http://www.ncbi.nlm.nih.gov/snp), Exome Variant Server (EVS), Seattle, WA, (http://evs.gs. washington.edu/EVS/), 6500 exomes and 1000 Genomes project (http://www.1000genomes.org/data).
Chromosomal hotspots
Pathway analysis of the 18 proteins
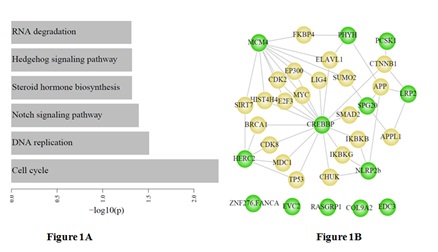
We constructed a subnetwork by mapping the 18 epilepsy-associated genes into the menthe protein interaction network, and found that eight of 18 epilepsy-associated genes showed indirect interactions between each other which were mediated by the important genes such as CREBBP (Figure 1B).
DISCUSSION
Epilepsy, defined by the presence of recurrent seizures, is associated with abnormalities in cognition, psychiatric status, and social-adaptive behaviors that are now referred to as neurobehavioral comorbidities. Given the increasing evidence of disease risk or causing genetic variants for epilepsy in non-Hispanic population, we hypothesized that the same and/or additional pathogenic mutations will be identified using a cutting edge technology, whole exome sequencing. With a stringent quality control and high coverage (126x) of this whole exome sequencing, we identified a total of 18 rare, heterozygous, predicted pathogenic variants that were present in at least one of the eight patients from a total of 11 patients, 9 of whom were of Latino decent.
The major finding of this study is that a number of potential disease causing mutations were identified. The current results provide pilot evidence that supports the CREBBP gene and Notch signaling pathway and cell cycle might be involved in pathophysiology of epilepsy. A recent study using an acute animal model also demonstrated a correlation between aberrant Notch signaling and epileptic seizures [24]. Furthermore, pathway analysis based on epilepsy associated SNPs identified by genome wide studies supports the cell cycle [25] in the disease pathophysiology. However, additional studies are warranted to examine the underlying mechanisms of these disease-causing mutations in epilepsy and confirm the findings in a large cohort in this unique population. As far as we know, this is the first report of pathogenic mutations identified for patients with epilepsy in the Latino population using whole exome sequencing (based on the PubMed Search on June 13, 2016).
Among 11 patients, patient #8 carrying six mutations had generalized epilepsy with occasional tonoclonic activity with urinary incontinence and postictal lethargy and confusion. EEG showed paroxysmal activity consistent with primary epilepsy and patient #12 carrying three mutations had a simple partial seizure with aura and MRI demonstrating encephalomalacia.
A number of newly identified disease mutations were also located in the “hotspots” previously reported in epilepsy related phenotypes, such as 6p21.3 microdeletions, where a Leu308Phe mutation in the CYP21A2 gene identified in the current study, were observed in more than four patients in different studies, including a most recent report [26]. The main feature of 6p21.3 deletion occurs in patients with developmental delay with severe speech impairment, seizures and behavioral abnormalities. The structure changes at 15q26.1 (the location of MEFE8 gene mutation observed in the current study) have been identified in a number of patients with epilepsy related phenotypes. A genome wide linkage meta-analysis mapped 19q13.42, (the location of the NLRP2-Glu292Asp mutation in the current study) to patients with genetic generalized epilepsy in 379 families [27]. Five studies reported 2q31.1 deletions in patients with seizure related phenotypes including a migrating partial seizure of infancy [28] and two patients with develop delay and seizure [29]. More than 30 children with 2q interstitial deletion have been reported (for review, please see [30]). Moreover, a recent study identified copy number variations in 16p13 in 60 patients with a combination of intellectual disability and genetic generalized epilepsy [31]. One of two studies [32,33] with positive linkage signals in lager families with epilepsy phenotypes also identified TBC1D24 mutation located at 16p13.3 in a family with epilepsy [32].
Furthermore, the KEGG pathway enrichment analysis (p<0.05) was performed for the 18 candidate proteins to illustrate the relationships between disease pathways of epilepsy and other pathways (Figure 1). The result from this analysis was shown that CREBBP protein was related to seven other proteins where mutations identified in patients with epilepsy.
We are aware of a number of limitations in this study. 1) We need to confirm these pathogenic mutations using a different approach which is our ongoing study: validating these 18 gene mutations using standard Sanger sequencing approach; 2) other variants, such as copy number variation, recent discoveries of putatively-causal structural abnormalities in epilepsy and epilepsy related traits, may not be captured using whole exome sequencing and 3) whole exome sequencing does not assess the impact of non-coding genome regions and whole genome sequencing is considered to the most comprehensive genetic test, although this may be hampered by challenges in data analysis and cost [34]. Therefore, future copy number variation analysis, whole genome sequencing and/or target gene sequencing will provide an opportunity for more in-depth molecular profiling of fundamental biological processes of the variants identified in the current study.
CONCLUSION
These first discoveries of functional genetic mutations using whole exome sequencing techniques provide insight into the susceptibility of the US Latino population with regard to epilepsy. These mutations represent important candidates for further investigation into the pathogenesis of epilepsy and may reveal potential drug targets for eventual therapy. Future studies will focus on how these functional mutations may influence the risk of epilepsy and confirm the findings in a large cohort and/or family study design.
ACKNOWLEDGMENT
We are grateful to all the families who participated in the study and many dedicated neurologists at TTUHSC and local hospitals at El Paso for help in patient ascertainment, particularly Dr. Richard D. Brower, a neurologist at TTUHSC_ElPaso for his patient ascertainment and helpful discussion.
Our thanks to Dr. Michael Escamilla, a professor at the TTUHSC-El Paso for allowing us to use his sample collection.
This study was supported, in part, by the TTUHSC Seed grant (PI, Dr. Xu) and TTUHSC SARP Mini grant (PI, Dr. Xu).
REFERENCES
- Banerjee PN, Filippi D, Allen Hauser W (2009) The descriptive epidemiology of epilepsy-a review. Epilepsy Res 85: 31-45.
- Corey LA, Pellock JM, Kjeldsen MJ, Nakken KO (2011) Importance of genetic factors in the occurrence of epilepsy syndrome type: a twin study. Epilepsy Res 97: 103-111.
- Poduri A, Lowenstein D (2011) Epilepsy genetics--past, present, and future. Curr Opin Genet Dev 21: 325-332.
- Veeramah KR, O’Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, et al. (2012) de novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet 90: 502-510.
- Science Daily (2013) New insight into the genetic causes of epilepsy. In: American Epilepsy Society, Science Daily, Rockville, MD, USA.
- Bras J, Guerreiro R, Hardy J (2012) Use of next-generation sequencing and other whole-genome strategies to dissect neurological disease. Nat Rev Neurosci 13: 453-464.
- Singleton AB (2011) Exome sequencing: a transformative technology. Lancet Neurol 10: 942-946.
- Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, et al. (2010) Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 42: 30-35.
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, et al. (2012) de novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485: 237-241.
- Michaud JL, Lachance M, Hamdan FF, Carmant L, Lortie A, et al. (2014) The genetic landscape of infantile spasms. Hum Mol Genet 23: 4846-4858.
- Suls A, Jaehn JA, Kecskés A, Weber Y, Weckhuysen S, et al. (2013) de novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet 93: 967-975.
- Epi4K Consortium; Epilepsy Phenome/Genome Project, Allen AS, Berkovic SF, Cossette P, Delanty N, et al. (2013) de novo mutations in epileptic encephalopathies. Nature 501: 217-221.
- O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, et al. (2011) Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 43: 585-589.
- Girard SL, Gauthier J, Noreau A, Xiong L, Zhou S, et al. (2011) Increased exonic de novo mutation rate in individuals with schizophrenia. Nat Genet 43: 860-863.
- Veltman JA, Brunner HG (2012) de novo mutations in human genetic disease. Nat Rev Genet 13: 565-575.
- Johansen Taber KA, Dickinson BD, Wilson M (2014) The promise and challenges of next-generation genome sequencing for clinical care. JAMA Intern Med 174: 275-280.
- [No authors listed] (1989) Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia 30: 389-399.
- Gonzalez S, Xu C, Ramirez M, Zavala J, Armas R, et al. (2013) Suggestive evidence for association between L-type voltage-gated calcium channel (CACNA1C) gene haplotypes and bipolar disorder in Latinos: a family-based association study. Bipolar Disord 15: 206-214.
- Sinha VK, Vaarties K, De Buck SS, Fenu LA, Nijsen M, et al. (2011) Towards a better prediction of peak concentration, volume of distribution and half-life after oral drug administration in man, using allometry. Clin Pharmacokinet 50: 307-318.
- Liu X, Jian X, Boerwinkle E (2013) dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat 34: 2393-2402.
- Li C, Li X, Miao Y, Wang Q, Jiang W, et al. (2009) SubpathwayMiner: a software package for flexible identification of pathways. Nucleic Acids Res 37: 131.
- Calderone A, Castagnoli L, Cesareni G (2013) Mentha: a resource for browsing integrated protein-interaction networks. Nat Methods 10: 690-691.
- Olsen ML, Sontheimer H (2008) Functional implications for Kir4.1 channels in glial biology: from K+ buffering to cell differentiation. J Neurochem 107: 589-601.
- Sha L, Wu X, Yao Y, Wen B, Feng J, et al. (2014) Notch signaling activation promotes seizure activity in temporal lobe epilepsy. Mol Neurobiol 49: 633-644.
- Bakir-Gungor B, Baykan B, Ugur ?seri S, Tuncer FN, Sezerman OU (2013) Identifying SNP targeted pathways in partial epilepsies with genome-wide association study data. Epilepsy Res 105: 92-102.
- Writzl K, Knegt AC (2013) 6p21.3 microdeletion involving the SYNGAP1 gene in a patient with intellectual disability, seizures, and severe speech impairment. Am J Med Genet A 161: 1682-1685.
- EPICURE Consortium, Leu C, de Kovel CG, Zara F, Striano P, et al. (2012) Genome-wide linkage meta-analysis identifies susceptibility loci at 2q34 and 13q31.3 for genetic generalized epilepsies. Epilepsia 53: 308-318.
- Carranza Rojo D, Hamiwka L, McMahon JM, Dibbens LM, Arsov T, et al. (2011) de novo SCN1A mutations in migrating partial seizures of infancy. Neurology 77: 380-383.
- Mitter D, Chiaie BD, Lüdecke HJ, Gillessen-Kaesbach G, Bohring A, et al. (2010) Genotype-phenotype correlation in eight new patients with a deletion encompassing 2q31.1. Am J Med Genet A 152A: 1213-1224.
- Pescucci C, Caselli R, Grosso S, Mencarelli MA, Mari F, et al. (2007) 2q24-q31 deletion: report of a case and review of the literature. Eur J Med Genet 50: 21-32.
- Mullen SA, Carvill GL, Bellows S, Bayly MA, Trucks H, et al. (2013) Copy number variants are frequent in genetic generalized epilepsy with intellectual disability. Neurology 81: 1507-1514.
- Falace A, Filipello F, La Padula V, Vanni N, Madia F, et al. (2010) TBC1D24, an ARF6-interacting protein, is mutated in familial infantile myoclonic epilepsy. Am J Hum Genet 87: 365-370.
- Palacio LG, Sánchez JL, Jiménez ME, Rivera-Valencia D, Jiménez-Ramírez I, et al. (2004) [Linkage analysis in an extended multigenerational family segregating for idiopathic epilepsy]. Rev Neurol 38: 916-920.
- Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, et al. (2014) Genome sequencing identifies major causes of severe intellectual disability. Nature 511: 344-347.
Citation: Villanos M, Mistrot JG, Ping YY, Ordonez J, Camarillo et al. (2016) Mutation Identification for Epilepsy in the US Hispanic Population Using Whole-Exome-Sequencing. J Cell Biol Cell Metab 3: 011.
Copyright: © 2016 Mariateresa Villanos, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.