Abbreviations
ALF: Acute Liver Failure
AIH: Autoimmune Hepatitis
ConA: Concanavalin A
GalN: D Galactosamine
HSCs: Hepatic Stellate Cells
KCs: Kupffer cells
LSECs: Liver Sinusoid Endothelial Cells
MDSCs: Myeloid Derived Suppressor Cells
MHC: Major Histocompatibility
ROS: Reactive Oxygen Species
SCID: Severe Combined Immunodeficiency Syndrome
TCR: T-cell Receptor
Key points
• Concanavalin A (ConA) induced T cell infiltration and liver injury is a model for acute hepatitis
• Important cell types include most white blood cells as well as stromal cells
• The ConA model has successfully been used to discover new pathways in hepatitis research
• New therapeutic interventions for hepatitis were discovered using the ConA model
Introduction
Autoimmune hepatitis
Autoimmune Hepatitis (AIH) is a chronic liver disease associated with raised plasma liver enzymes like transaminases and the presence of autoantibodies [1,2]. The initial trigger of the disease is unknown but different genetic risk factors have been identified [3,4]. In general, it is believed that multiple perturbations, unidentified environmental factors or drugs and the loss of self-tolerance are needed for the onset and development of AIH. The mechanisms for loss of self-tolerance leading to auto reactivity remain unclear but major contributors such as impaired thymic negative selection and expansion of promiscuous T cell clones are suggested [5]. In AIH, presentation of a self-antigen peptide within a Major Histocompatibility (MHC) II molecule by professional Antigen Presenting Cells (APCs) initiates liver damage. This antigen presentation, in combination with exposure to various cytokines, drives the differentiation of uncommitted CD4 helper T cells and immune reaction that is skewed to a Th1 and Th17 response. However, the Th2 response cannot be ignored because it is important in the production of auto-antibodies, which trigger cellular cytotoxicity and complement activation [6,7]. The current standard treatment of AIH patients consists of non-specific immune suppressing drugs such as corticosteroids in combination with azathioprine, which leads to a decrease in serum aminotransferase levels [8].
AIH animal models
Despite extensive research on the pathogenesis of AIH, the details of the mechanism remain elusive and the current treatments are non-specific and insufficient. The need for faithful and reliable animal models is therefore high. However, finding a suitable animal model is difficult due to the fact that AIH lacks a precise time of onset, etiological agent and the clinical phenotype is variable. According to Czaja et al., the current AIH models can be divided in two categories: the pathogenic and therapeutic models [1]. Pathogenic models focus on characterization of individual mechanisms contributing to occurrence and severity of the disease. In these perturbed models, individual pathways can be isolated and manipulated in a genetic homogenous background. Next to transgenic models in which target antigens are expressed under the control of liver-specific promoters, several inducible models such as the D Galactosamine (GalN) in combination with lipopolysaccharide/enterotoxin hepatitis, and Concanavalin A (ConA) liver injury have been defined [9]. Despite the insights that can be gained with the pathogenic models, therapeutic models are needed to test new molecular interventions. These models require a consistent aggressive phenotype and close similarity to human disease. One of the major challenges in creating these models is the difficulty to lose self-tolerance due to the tolerogenic nature of the liver. In order to break this inherent tolerance, a preconditioning of the liver with an adjuvants or stimulators of the innate immune response might be needed. Recently, a new conditional knockout model deleting TNF Receptor Associated Factor (TRAF) 6 in medullary thymic epithelial cells and a transgenic CYP2D6 model using a natural human autoantigen to induce a long-term auto-immune damage to the liver were identified [10-12]. For a more elaborate overview of AIH animal models we refer to [1,9,13,14]. In this review we will focus on the ConA model of hepatitis as a well-established model to investigate T cell and macrophage dependent liver injury.
ConA induced liver injury
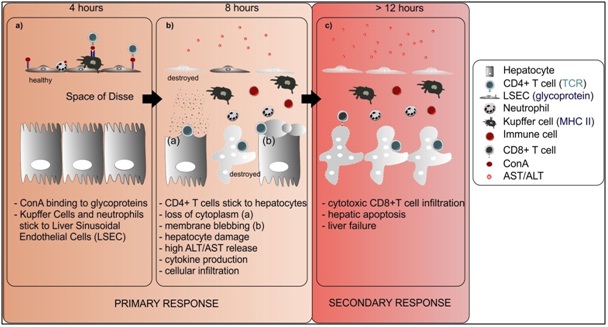
ConA is a plant lectin purified from Canavlia brasiliensis [15]. Lectins are proteins that bind to sugar residues on the surface of a wide variety of cell types, leading to agglutination of cells, mitogenic stimulation, or cytotoxicity of lymphocytes and macrophages [16]. ConA binds to the mannose receptor on Liver Sinusoidal Endothelial Cells (LSECs) early after administration (4 hours), leading to the breakdown of their membrane and subsequent cell death. At this time point, a pronounced sticking of leukocytes to the hepatic endothelium is also observed. Because of the detachment of LSECs, the binding of ConA to Kupffer Cells (KCs) and neutrophils is facilitated. Four hours later, CD4 + T lymphocytes are found attached to hepatocytes and are able to recognize MHC II molecules bound with ConA, leading to their activation. Eight hours after ConA, the sinusoidal parts of the hepatocytes show severe damage with ruptures of the cell membrane, membrane blebbing and loss of cytoplasm. The ConA model is mainly characterized by a Th1 response leading to extensive inflammation by elevated cytokine levels. Infiltration by cytotoxic CD8+ T cells is a secondary event that results in hepatic apoptosis and eventually liver failure [17] (Figure 1). Intravenous ConA injection leads to an early hepatic cytokine storm with antigen independent activation and T cell proliferation. This results in severe liver damage by necrotic cell death of hepatocytes and release of transaminases Alanine Transaminase (ALT) and Aspartate Transaminase (AST) into the blood. Histopathological examination of the other major organs shows no obvious signs of organ injury except in the liver [17]. The ConA model is suitable for investigation of the involvement of T cell infiltration but also to study cytokines produced by T cells such as Tumor Necrosis Factor (TNF) and Interferon-Gamma (IFN-γ) [18].
Despite the above differences between the ConA model and human AIH, this model is considered as one of the best models for mimicking and studying certain mechanisms of AIH, and it has played an important role in AIH drug development [9]. However, decades of ConA-induced hepatitis research demonstrate that the model suffers from substantial variability resulting in inconsistency of reported results. The most important factors to take into account when using this model are the dose, genetic background, gender and age of the mice and microbial status of the animal facility. For more details we refer to a recently published standard operating procedure [18]. In this review, we present an overview of the current understanding of the cell types (Table 1) and molecules which are involved in ConA-induced liver injury. In addition, we will summarize successful therapeutic interventions and their effects on important factors and cell types in disease development.
T Cells
T cell activation has direct cytotoxic effects on hepatocytes in vitro [69] and is crucial in the development of liver damage caused by ConA stimulation in vivo [17]. The necessity of T cells for ConA-induced liver injury was documented by different groups using a variety of tools such as mice with Severe Combined Immunodeficiency Syndrome (SCID) lacking immunocompetent T and B cell [17], athymic nude mice with functionally defective immature T cells [17,70], RAG-1 deficient mice devoid of T and B cells [19,71] and a Th1.2 antibody that depletes all antigen-bearing T cells [17]. More specifically, the induction of liver injury requires CD4+ T cells, not CD8+ T cells, because depletion of CD4+ T cells with a monoclonal antibody against CD4 glycoprotein but not against CD8 completely protected against ConA-induced liver injury [17, 26,71]. However, depletion of CD4+ and CD8+ reduced liver injury to a greater extent than depleting only CD4+ T cells [26]. The dominant role of CD4+ T cells in ConA hepatitis is no surprise knowing that, in early reports, administration of ConA to mice and rats led to the accumulation of CD4+ cells in the liver, but not CD8+ T cells [72,73]. Conversely, other reports did observe CD8+ T cells accumulation in the liver [74] and these cells were even the main producers of IFN-γ [45]. Moreover, in vitro data show that ConA induces more proliferation of CD8+ T cells than CD4+ T cells [75]. These results indicate that ConA affects CD8+ T cells but the cytotoxic role of CD8+ T cells is secondary to the role of CD4+ T cells [74].
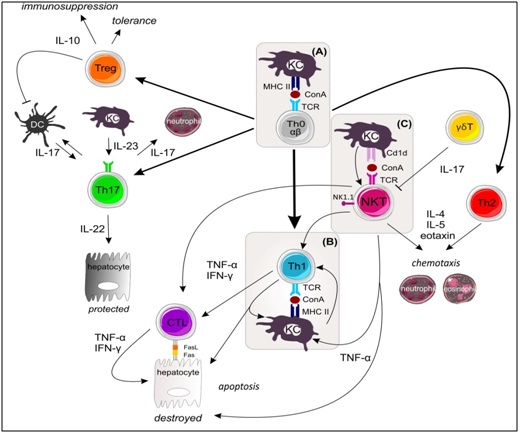
ConA activates T cells to proliferate, produce cytokines and migrate to the liver [46]. Several adhesion molecules play an important role in accumulation of CD4+ T cells but not CD8+ T cells [76]. By helping APCs and cytotoxic CD8+ T cells, CD4+ helper T cells are important in the initiation of an adaptive immune response. CD4+ T cells are divided into four major subsets based on their secreted cytokines and expression profile of transcription factors, namely, Th1, Th2, Th17 and regulatory T cells (Tregs) [77]. The involvement of and interplay between these T cell subsets in ConA-induced liver injury will be discussed in the following sections and is summarized in Figure 2.
Th1 cells
The differentiation of Th0 to Th1 cells is mainly induced by Interleukin (IL)-12 and can be further enhanced by IFN-γ in a feedback loop, because next to TNF-α and IL-2, IFN-γ is one of the major cytokines secreted by Th1 cells [78,79]. Several studies using recombinant IFN-γ, IFN-γ antibodies and deficiency have shown that IFN-γ plays a crucial role in the development of ConA-induced liver injury [27,37,47, 80-82]. In the absence of IFN-γ, hepatocytes don’t die by apoptosis and production of TNF-α by activated macrophages is reduced [79-81,83]. Another key Th1 associated cytokine is TNF-α, which is also local and systemically strongly induced upon ConA administration [81]. However, the importance of this cytokine in the development of liver injury has been controversial. Initial reports demonstrate that neutralization of TNF-α was unable to prevent the development of ConA-induced hepatitis [17,81]. Using mice deficient in TNF Receptor (TNFR) 1 or TNFR2 deficient mice and TNF-α neutralizing antibodies, later studies reported protection against ConA-induced liver injury [37,47,84-86]. Interestingly, cooperative signaling between the two TNFRs is important for the induction of liver damage [80]. TNFR2 on the parenchyma but not on leukocytes can attract adaptor molecules such as TRAF2 which facilitates TNFR1 induced activation of the pro-apoptotic pathway of nuclear factor NF-κB [87,88]. TNFR1, on the other hand, is necessary for NF-κB activation in immune cells [86].
IL-12 is produced mainly by monocytes, macrophages and Dendritic Cells (DCs) [89] and plays an important role in the production of Th1 cytokines as well as Th2 cytokines by activation of Signal transducer and activator of transcription (STAT)4 [90,91]. IL-12 is detrimental in ConA-induced liver injury model as its administration exacerbates the signs of ConA-induced hepatitis. Additionally, its neutralization ameliorates them by reducing the expression of Th1 and Th2 cytokines [82,92]. In contrast, recent results using IL12 and STAT4 deficient mice reveal a protective role of IL12 induced activation of STAT-4 via indirect or direct down regulation of FasL in Natural Killer T (NKT) cells. These conflicting results indicate that IL-12 and STAT4 may play a dual role in ConA-induced hepatitis by promoting cytokine production and inhibiting NKT cell function [93].
Th17 cells
The combination of Transforming Growth Factor (TGF)-β, IL-6 and IL-21 is needed for the differentiation of Th17 cells, which are characterized by the production of IL-17, IL-21, IL-22 and TNF-α [96,97]. After triggering Th17 cell differentiation, the cells start to produce IL-21, which causes amplification by positive feedback and IL-23 receptor expression. Production of IL-23 and IL-21 induces the expression of RAR-related orphan receptor (ROR)γt, which is necessary for the development of Th17 cells [98]. Development of Th17 is suppressed by IFN-γ and IL-4, which commit Th0 cells to Th1 and Th2, respectively. Next to RORγt, Th17 cells activate STAT3 and RORα transcription factors, leading to the production of IL22, IL-17 A and IL-17F [99]. IL-17 family members play important roles in local tissue inflammation and cause KCs to release pro-inflammatory cytokines such as IL-6 and TNF-α but also influence neutrophil recruitment/activation and Th1/Th2 activation [100]. IL-17 is believed to be a marker of AIH severity. In the ConA model, increased expression of IL-17 was also observed in CD4+ T and NKT cells [100,101]. Moreover, IL-17 secretion by activated T cells upon ConA stimulation promoted TNF-α and IL-6 production by KCs, causing liver damage [100]. Supporting this pro-inflammatory role of IL-17, neutralization or deficiency of IL-17 led to Reduce ConA-induced liver damage whereas exogenous IL-17 aggravated it [71,100,102,103]. Nevertheless, the complete role of IL-17 in the pathogenesis of ConA-induced hepatitis remains unclear since an immunosuppressive role was recently described [104]. By promoting inducible Nitric Oxide Synthase (iNOS) expression in mesenchymal stem cells and induction of cytokine/chemokine gene expression, IL-17 can enhance in vivo immunosuppressive effects in ConA-induced hepatitis [103-106].
IL-22 is known to protect hepatocytes from the destructive effect of IFN-γ produced by different immune cells [105]. IL-22 has no direct effect on immune cells but affects liver tissue by activating STAT3 and Akt in hepatocytes, leading to the induction of anti-apoptotic and cell survival genes. The hepatoprotective role of IL-22 in ConA-induced injury was confirmed by treating IL-22 deficient mice [105]. It is important to note that Th17 cells are not the only source of IL-22, for example, Notch signaling in CD4+ T cells is crucial for IL-22 expression in a Th17 independent manner [107]. Interestingly, IL-22 seems to be pro-inflammatory in the presence of IL-17 but protective in its absence [108]. IL-23 does not initiate Th17 cell differentiation but acts as a survival cytokine maintaining Th17 responses. Th17 master regulators STAT3 and RORγt are essential for the expression of its receptor [109]. In ConA-induced hepatitis, the induction of IL-23 expression and Notch activation results in the production of IL-22 which is critical for protection. IL-23 induces the expression of IL-17 as well, which is detrimental in the pathogenesis, showing that IL-23 has both a hepatotoxic and a protective role [103].
As mentioned above, STAT3 is an important transcription factor involved in the induction of the Th17 specific cytokine profile. It has opposing roles in myeloid and T cells in ConA-induced hepatitis. In myeloid cells it inhibits STAT1 and NF-κB activation; thereby inhibiting hepatitis by downregulating the production of various cytokines such as IL-6, IL-12 and IFN-γ. In T cells, it leads to the expression of Th17 specific inflammatory cytokines, including IL-17, which might play a detrimental role in ConA-induced liver injury [102].
Regulatory T cells (Tregs)
The main function of CD4 regulatory T cells (Tregs) is to suppress the activation and/or effector actions of immune cells such as DCs, T, B, NK and NKT cells. Tregs are divided into three subsets: CD4+ CD25+ Foxp3+ cells, Tr1 cells and Th3 cells. Resident CD4+ CD25+ Tregs exert their suppressive function by direct cell contact via membrane bound molecules. This type of Tregs develops directly from CD4+ T cell precursors during positive selection. In contrast, induced Tr1 and Th3 cells do not use contact but secrete IL-10 or TGF-β and differentiate from naive conventional CD4+ T cells [110,111]. In ConA-induced hepatitis, the absolute number of Foxp3 positive Tregs is significantly increased and CD4+ CD25+ Treg activities/functions are enhanced [61,112]. Tregs are attracted to the liver by LSECs and hepatocytes expressing C-X-C motif chemokine ligand (CXCL) 9/10/11 leading to attraction of C-X-C Receptor (CXCR)3 positive CD4+ T cells and Tregs [113,114].
The inhibitory function of Tregs is mediated by TGF-β, which is needed to suppress the injury caused by effector T cells [61]. Depleting CD25+ cells aggravated ConA-induced injury, and treating mice with CD4+ CD25+ Tregs made them less susceptible; this illustrates the therapeutic potential of Tregs [62]. For example, galectin-9 plays an important role in inducing apoptosis of TIM-3 positive activated CD4+ T cells and is predominantly expressed on Tregs [115-117]. In ConA-induced hepatitis, TIM3 neutralization, which blocks the effects of the endogenous ligand of galectin-9, aggravates liver injury by inhibiting the suppressive activities of Tregs [26,118]. Moreover, administration of galectin-9 ameliorates injury by downregulating the number of CD4+ effector T cells and their infiltration and cytokine production in the liver [26]. This example shows that the number of Tregs is important in overcoming ConA-induced hepatitis and interfering with Treg amount either exacerbates or improves injury.
ConA also induces Tr1 cells which secrete IL-10 and are Foxp3 negative [63]. IL-10 produced by Tregs and KCs plays a prominent role in the induction of tolerance. Interestingly, one single sublethal dose of ConA can induce tolerance within eight days. This tolerogenic state is characterized by an anti-inflammatory cytokine profile, with IL-10 as one of the major cytokines [62]. Restoring self-tolerance could be a valid therapeutic strategy because deficiency of Treg number or function plays a key role in ConA induced hepatitis. For a detailed review on using regulatory T cells to treat liver disease we refer to [119]. However, the role of Tregs in the pathogenesis of AIH and other autoimmune diseases is not clear. Recent reports show that Treg frequency and function are not impaired in AIH but the liver infiltrating Tregs are not able to block disease progression [120].
γδ T cells
In humans, 95% of the T cells express a T Cell Receptor (TCR) consisting of α- and a β-chain (αβ T cells), whereas in 5% of T cells the TCR has both γ- and δ- chains (γδ T cells). The features and functions of γδ T cells are distinct from those of αβ T cells, and γδ T cells recognize both non-self and self-antigens [121]. γδ T cells can be divided into two major subsets: antigen experienced γδ T cell produce IFN-γ whereas antigen naive γδ T cells produce IL-17 [122]. According to their TCR, peripheral γδ T cells are divided into Vγ1 γδ T and Vγ4 γδ T cells [123-125]. Vγ4 γδ T cells are the main source of IL-17 in the periphery and play a protective role in ConA-induced hepatitis by negatively regulating IFN-γ production by NKT cells [59]. In contrast, Vγ1 γδ T cells were not protective, supporting the idea that these γδ T cell subsets have divergent functions in some disease models [126].
NKT Cells
NKT cells are a special subset of lymphocytes that express both the TCR with the invariant VαJα-chain and NK cell associated markers, including NK1. 1. These cells are considered to form a bridge between the innate and the adaptive immune systems. NKT cells are enriched in the liver and constitute about 30% of hepatic lymphocytes [127]. There are two distinct subpopulations of NKT cells: type I (95% of NKT cells) and type II (5% of NKT cells) [128]. Type I invariant iNKT cells have a highly restricted TCR repertoire, in contrast to the type II non-invariant NKT cells, which express a wide range of TCRs [129,130]. The crucial role of NKT cells in the pathogenesis of ConA-induced hepatitis has been shown by different studies using NKT deficient mice, depleting antibodies or NOD mice partially iNKT deficient [19,37,49,50,51,92,131]. Although NKT cells are one of the predominant effector cells in ConA-induced liver injury, they are rapidly depleted from the liver by Fas/FasL mediated apoptosis [49].
Activation of NKT cells is mediated by presentation of lipid antigens by CD1d but needs additional signals from KCs, LSECs, Hepatic Stellate Cells (HSCs), Myeloid Derived Suppressor Cells (MDSCs) and hepatocytes [132]. One of the most important signals is IL-12, produced mainly by APCs, which acts on iNKT via the IL-12R and triggers increased production of IL-4 [92,133]. In ConA-induced hepatitis, NKT cells are required for IL-4 induction which is essential for TNF-α production and liver damage [51]. Once they are activated, NKT cells release large amounts of Th1 cytokines (IFN-γ, TNF-α and IL-2) and Th2 cytokines (IL-4, IL-5, IL-10 and IL-13) [130]. NKT cells directly induce hepatocyte death through release of TNF-α or perforins/granzyme B and/or upregulation of FasL [134]. Next to this, upon TCR engagement iNKT cells can rapidly produce a mixture of cytokines involved in the differentiation of Th0 cell into Th1 or Th2 cells [135-137]. So by releasing of IFN-γ, NKT cells can indirectly induce hepatocyte death by driving a Th1 biased phenotype and converting CD8+ T lymphocytes into cytotoxic T lymphocytes (CTLs). (Figure2) It is clear that NKT cells play a well-established detrimental role in the course of ConA-induced injury. Hence, interfering with the development, recruitment or activation of NKT cells in the liver could be an effective therapeutic strategy.
Hepatocytes
Upon ConA challenge, IL-1β serum levels are increased, and this cytokine is a key mediator in the pathogenesis of the disease [84,85]. Hepatocytes are the major source of both circulating and hepatic IL-1Rα, which plays a critical role in the resolution of hepatic damage [138]. After ConA stimulation, TNF-α induces liver damage by activating both TNFR1 and TNFR2 [80,139]. However, TNF-α is not the only factor inducing apoptosis in the liver. Hepatocytes are very prone to Fas/FasL-mediated apoptosis because of their strong expression of Fas [140]. This type I integral membrane protein of the TNF receptor family transduces cell death signals and administration of a monoclonal agonistic Fas stimulating antibody leads to massive hepatocyte apoptosis and consequent liver damage [141-143]. However, the role of Fas/FasL in ConA-induced hepatitis remains controversial. Several studies have shown that blocking Fas/FasL signaling using antibodies, deficient mice or siRNA prevents ConA-induced liver injury [81,134,144,145]. In contrast, others researchers did not demonstrate a significant role for Fas/FasL but claimed that perforin-mediated cytotoxicity is important [69,85,134,146]. Interestingly, deficiency of caspase 1, which is a common intermediate of TNF-α and FasL signaling, also leads to protection against ConA-induced hepatitis [85,147].
As mentioned above, IL-22 targets epithelial cells such as hepatocytes and plays an essential antihepatotoxic role in ConA-induced injury [105,148-150]. The protective role of IL-22 is mediated through activation of STAT3 and subsequent induction of the anti-apoptotic proteins Bcl2 and Bcl-xL [148,149]. Next to this, strong IL-33 expression in hepatocytes is controlled by NKT cell activation [50]. IL-33, a new member of the IL-1 family, reduces the severity of liver damage caused by ConA stimulation by recruiting regulatory T cells to the liver. The expression of IL-33 is dependent on TNF-Related Apoptosis-Inducing Ligand (TRAIL) but not on TNF-α and FasL [151]. TRAIL induces apoptosis by binding to death receptors, but it also initiates necroptosis [152,153]. TRAIL induces necroptosis by activating PARP-1 in a RIPK1/3 dependent manner, which leads to depletion of ATP and subsequently to programmed necrosis [154]. Since TRAIL is a crucial mediator of ConA-induced liver injury, this type of damage is driven not only by apoptosis but also by necroptosis [155]. Furthermore, elevated RIPK1 and RIPK3 expression indicate that necroptosis occurs during pathogenesis [156].
Myeloid-Derived Suppressor Cells
Myeloid Derived Suppressor Cells (MDSCs) expand during cancer, infection and inflammation, and have an outstanding ability to suppress T cell responses. This heterogeneous pool of myeloid cells has the morphology of granulocytes and monocytes [157]. Murine MDSCs are characterized by surface co-expression of Gr-1 and CD11b and are further subdivided into two major populations: CD11b+ Ly6G+ Ly6Clow granulocytic MDSCs and CD11b+ Ly6G- Ly6Chigh monocytic MDSCs [158]. MDSCs use various mechanisms to influence both innate and adaptive immune responses. Most importantly, they deprive T cells of L-cysteine and L-arginine, generate oxidative stress, and can activate or expand Tregs (for a review on MDSC functions we refer to 159,160). To facilitate the suppression of T cell responses, MDSCs need iNOS, Arginase 1 and Reactive Oxygen Species (ROS) [161]. Recent findings suggest that the accumulation of MDSCs is linked to different types of inflammatory diseases such as inflammatory bowel disease [162] but also hepatitis B and C infections [66,163-165]. Moreover, in ConA-induced liver injury an expansion of MDSCs in the liver was observed and these cells suppressed the proliferation of an early T cell subset population [20].
Different therapeutic interventions have been shown to be associated with the induction and/or expansion of MDSCs during ConA-induced liver inflammation [20,66,67]. For example, treatment with glucocorticoids, which are strong anti-inflammatory molecules working through the GC receptor GR, led to accumulation of MDSCs and protected against liver injury [20,166]. A specific liver MDSC population with an unique surface marker expression profile was recently identified. Moreover, Zhu et al., demonstrated that only this population could inhibit CD4+ T cell responses and this inhibitory capacity depends on de novo expression of iNOS and subsequent NO production upon CD4+ T cell contact [65]. In human AIH patients, evidence for the presence of MDSCs is limited [167]. However, accumulation of CD11b + cells in liver biopsies has been reported [168]. Data on MDSC function in mouse models of AIH show that exhaustion of MDSCs and impairment of MDSC effector functions are part of the pathogenic mechanisms of the disease. Restoring the MDSC suppressive capacity or increasing the accumulation of MDSCs could thus be a promising therapeutic strategy for AIH. However, specific methods to generate these cells ex vivo so that they will be suitable for administration to patients will be challenging [159]. Especially because MDSCs are not terminally differentiated cells so depending on their surrounding milieu, they may mature into other myeloid cell types.
Natural Killer Cells
The cytotoxic and cytokine response of Natural Killer (NK) cells depends on their interaction with other cells of the immune system, such as T cells and NKT cells, as well on the cytokine environment [169]. In ConA-induced hepatitis, large numbers of NK cells are recruited from the bone marrow and spleen to the liver. Activation of NK cells requires other types of immune cells, such as NKT cells and T cells, and depends strongly on IFN-γ production [58]. Since NK cells need other cells in order to perform their function, it could be expected that in vivo depletion of NK cells alone does not ameliorate ConA-induced liver injury [36,51,58,170].
Kupffer cells
The liver resident macrophages or KCs constitute the first line of defense in the liver sinusoids that is in direct contact with all kinds of danger signals, such as microbial debris and gut derived pathogenic microbes. KCs phagocytose this exogenous material and degrade it, but they are also an important source of various inflammatory mediators. During disease, KCs interact with different neighboring cell types and the mediators they produce are central in influencing these cells. For a more detailed overview of the different cell-cell interactions and macrophage associated mediators in liver injury, we refer to Yang et al., [171].
Macrophages differentiate into M1 or M2 subsets, which are characterized by distinct markers and functions. The switch between pro-inflammatory M1 and anti-inflammatory M2 macrophages is controlled by mediators in the micro-environment of macrophages. A shift in the M1-M2 balance is correlated with the state of inflammation [171]. At the onset and during the first phase of acute hepatitis, macrophages exhibit a classical M1 phenotype and are hepatotoxic. In this phase, ConA activates KCs to produce large amounts of pro-inflammatory cytokines and promotes liver injury [80,172]. The essential role of KCs in disease development has been demonstrated by various research groups, who showed that KC depletion represses liver injury [38-40, 87]. Not surprisingly, depleting KCs does not affect the ability of ConA to induce the expression of important cytokines such as TNF-α and IFN-γ, suggesting that KCs are not the main producers of these factors. In contrast, the production and release of superoxide ROS is reduced after KC depletion; these species are critical in ConA-induced hepatitis [37,39]. In the second phase of hepatitis, macrophages display an M2-like phenotype, which is characterized by the production of anti-inflammatory and regenerative cytokines such as IL-10, TGF-β and IL-6 [171]. M2 macrophages counteract the overwhelming pro-inflammatory events during the resolution phase of ConA-induced hepatitis [173]. The most important mediators involved in suppressing inflammation and promoting tissue repair have been reviewed by Yang et al., and will not be discussed further [171]. In a third phase, six hours after ConA challenge, the total number of KCs decreases but the remaining KCs have a more potent pro-inflammatory phenotype characterized by high IL-1β secretion [174]. As mentioned before, IL-1β is known as a critical pro-inflammatory cytokine during liver injury [175].
Neutrophils
Neutrophils are the first cell type to arrive at the site of inflammation in response to chemotactic factors. They migrate from the bloodstream to their target, where they attempt to resolve inflammation [78,176,177]. ConA challenge induces a recruitment of neutrophils to the liver within four hours. ConA can bind and activate neutrophils with consequent expression of adhesion molecules and release of reactive oxidants [179]. Accumulation of neutrophils is seen in the necrotic areas in the liver [47,48]. The activation and recruitment of neutrophils is a crucial step in the pathogenesis of ConA-induced liver inflammation [45,46]. Depleting neutrophils with an anti Gr1 antibody does not completely prevent but represses liver injury and leukocyte trafficking. Moreover, Bonder et al., showed that without neutrophils ConA cannot induce the recruitment of T cells and subsequent liver injury [46]. Activation of neutrophils leads to the expression of various mediators, such as IFN-γ, TNF-α and IL-4, which are known crucial factors in the development of ConA-induced liver injury, as discussed before [45].
Eosinophils
Eosinophils are important in mediating tissue damage through the release of toxic granule proteins, pro-inflammatory cytokines, chemokines and lipids. The activation of NKT cells in ConA-induced hepatitis results in a strong IL-4/STAT-6 dependent production of eotaxin and IL-5, leading to eosinophil and neutrophil infiltration into the liver [95]. IL-5 is not only responsible for the maturation, proliferation and differentiation of eosinophils but also, together with eotaxin, for the recruitment of eosinophils to the liver. The importance of eosinophils in ConA-induced liver inflammation was shown using the CCR3 antibody to deplete eosinophils. Treatment with this antibody and neutralization of IL-5 led to protection against ConA-induced injury [48]. Next to IL-5, Smad3 is an essential mediator of TGF-β signaling and is important for the viability of eosinophils in vitro, supporting the idea that upon ConA stimulation, Smad3 promotes eosinophil function and survival [180,181].
Dendritic Cells
In contrast to conventional DCs (CD11c+CD11b+ B220-) present in the spleen and other organs, hepatic DCs are thought to be plasmacytoid (CD11c+CD11b- B220+) [182]. Hepatic DCs are weak activators of immune responses and are believed to be more immunosuppressive and to have a somewhat immunotolerance phenotype [183,184]. During the early stages after ConA challenge (3-6h), the absolute number of DCs in the liver decreases, but during the later stages (12-24h) their number increases again. Using adoptive transfers, Tomiyama et al., showed that only hepatic DCs could suppress the hepatic damage induced by ConA and this might be related to their ability to suppress IL-12 secretion and the subsequent Th1 response [184].
Stromal Cells
Stromal cells lining the hepatic sinusoids are the first cells to encounter gut-derived or systemic antigens. There are two major types, both of which can modulate the immune response: Liver Sinusoidal Endothelial Cells (LSECs) and Hepatic Stellate Cells (HSCs).
LSECs are the most abundant non parenchymal cell population in the liver. They are unique endothelial cells that line the hepatic sinusoids and define the space of Disse. Although LSECs express low levels of MHC II and co-stimulatory molecules, they are scavengers that compete with DCs for the uptake of antigens circulating in the liver [185]. By physically interacting with DCs, LSECs can reduce the antigen presenting function of DCs [186]. After physical contact, DCs lose their ability to prime naive T cells due to reduced expression of co-stimulatory molecules. LSECS can also promote CD4+ T cell migration in a chemokine dependent way [187]. It was recently shown that the liver sinusoidal endothelium contains an intracellular transport system for chemokines to induce local recruitment of circulating T cells. During ConA-induced inflammation, LSECs internalize CXCL12 via CXCR4 mediated endocytosis, leading to enhanced CD4+ T cell migration [188]. LSECs can also reduce CD4+ T cell activity and promote the induction of Tregs using membrane bound TGF-β [114,189,190].
HSCs are present in the perisinusoidal space between LSECs and hepatocytes. In normal conditions, HSCs (Ito cells) are important for Vitamin A storage and for regulation of blood flow in the sinusoids. In inflammatory conditions, HSCs form a second line of defense by limiting the effector function of T cells that have extravasated and thereby they prevent tissue damage. HSCs produce large amounts of anti-inflammatory mediators like IL-10 and TGF-β [191]. By doing so, they can suppress DC function [192]. In addition, HSCs can induce NKT cell response by producing IL-15, present peptides to CD4 and CD8+ T cells, and induce T cell priming [193]. Next to LSECs, HSCs are the second APCs that reside in the liver. HSCs also facilitate the differentiation of inflammatory monocytes into MDSCs, which impair T cell function and proliferation [194]. Moreover, HSCs can induce and expand Tregs but the mechanism is not known [195,196]. For a full overview of hepatic immune regulation by stromal cells we refer to Schildberg et al., [197].
Therapeutic Interventions
Because of the great amount of cell types and mediators involved in the pathogenesis of the ConA induced liver inflammation; a lot of different therapeutic strategies have been tested in the last decades (depicted in the last column of Table 1). In most studies, strategies which inhibit T cell and macrophage activation leading to reduced cytokine production and liver damage are used. Known immunomodulatory agents like corticosteroids, cyclosporine A, paeoniflorin, glucocerebroside, curcumin and flavonoids -quercetin, hesperidin and baicalein- were able to reduce ConA associated cytokine production [17,22,43,53,119,198,200]. Unfortunately, most of the reported therapies are only effective when administered prophylactically (marked as [P] in table 1) and some even lose their potential when used therapeutically (marked as [T] in table 1). For example, pentoxyfilline, a strong suppressor of TNF-α release, is able to rescue the phenotype when administered prophylactically but the effect was lost when used therapeutically [33,201]. Luckily, there have been some studies identifying valid therapeutic strategies. For example, Y-40138 significantly suppressed the development of ConA-induced hepatitis by suppression of TNF-α, MIP-2, IL-4 and IFN-γ production [201]. Additionally, both quercetin and TP58, a novel thienopyridine derivative protect against liver injury by inhibiting NF-κB mediated inflammation [41,198]. Other strategies targeting regulatory T cells and/or NKT cell function have also been described. For example, oral administration of DT56a, a natural product isolated from soybeans, redistributed the CD4+ CD25+ T cells resulting in less liver damage [57]. New and interesting therapeutic opportunities arose when immunosuppressive MDSCs were shown to be induced upon ConA stimulation [20]. Later studies using dexamethasone show that its positive effect is specifically due to an increased MDSC induction [20]. Next to this, cannabidiol, a non-psychoactive cannabinoid and IL-25 treatment can reduce ConA induced inflammation and protect mice from liver injury by triggering the induction of MDSCs, opening a new window for therapeutic strategies [66,67].
Conclusion
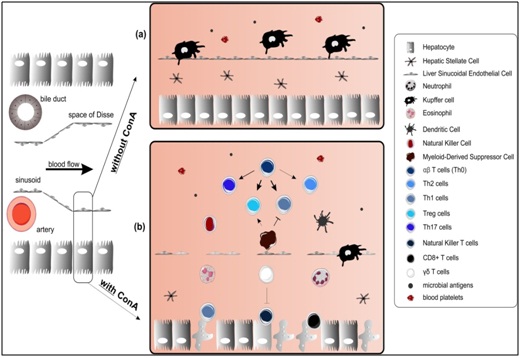
The ConA model mimics important features of acute liver injury. The immune activation/infiltration and the involvement of T cells in this model make it particularly relevant to study acute liver injury. In this review we presented an overview of the different cell types and their mediators driving ConA-induced hepatitis. Decades of research have shown that the pathogenesis is more complex than anticipated by Tiegs et al., in 1992 (shown in figure 1) [17]. The great variety of immune cells, hepatocytes and stromal cells that play crucial roles in the development of the disease and the fact that different cell types influence the effector functions of their neighboring cells by producing different cytokines, chemokines and other effector molecules (shown in Figure 2) are responsible for this high complexity. It is now clear that ConA-induced hepatitis is the result of a well-orchestrated interaction network (Figure 3) and novel mediators are still being identified meaning that the full complexity of pathogenesis is not yet understood. Fortunately, successful therapeutic strategies targeting a specific mediator or cell type have been already identified. However, recent findings, like the induction of MDSCs, have great therapeutic potential but have not been investigated to a great extent and should be focused on in the future.