Molecular Insights on the Interactions of Nitrosamines from Cigarette Smoking with CYP2A13 using Molecular Docking and Molecular Dynamics Simulation
*Corresponding Author(s):
Jorddy N CruzFaculty Of Pharmaceutical Sciences, Laboratory Of Preparation And Computation Of Nanomaterials, University Of The Amazon, Belém, Brazil
Tel:+55 66065219,
Email:jorddynevescruz@gmail.com
Abstract
Keywords
INTRODUCTION
Another serious illness that affects most smokers is cancer [5], especially lung cancer. This disease may be associated with high frequency of cigarette smoking as well as its prolonged use during life [6,7]. Increased incidences of lung cancer have been reported since the 1930s, and by 2012 it was estimated to have been responsible for the death of approximately 1,590,000 people. It remains one of leading causes for cancer deaths globally. Among men, the deaths occur mainly in Central and Eastern Europe and among women in North America, Northern Europe, Eastern Asia, Western Europe, Oceania and the Caribbean [8,9]. Overall, the mortality rate due to lung cancer is the highest in countries where people start smoking earlier, such as in North America and Europe [10].
Figure 1 shows the chemical compounds N'-Nitrosoanabasine (NAB) and N'-Nitrosoanatabine (NAT) formed from anabasine and anatabine, respectively [11]. One of the likely causes of lung cancer is the nitrosamines derived from tobacco smoke [12].
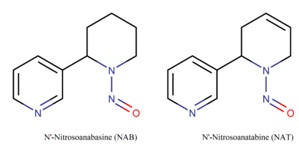
Figure 1: 2D structural formulas of nitrosamines NAB and NAT.
The molecules present in tobacco and tobacco smoke begin to act in the body only after enzymatic activation that can be achieved by cytochrome P450 2A13 (P450 or CYP) and is considered a key reaction pathway for initiation of bioactivation of carcinogenesis, and consequently of the progressive changes in the biological profile of the cell [13,14].A useful tool to demonstrate interactions between nitrosamine linkers and CYP2A catalytic sites is the molecular docking study, which comprises the mechanism of activation and/or oxidation of substrates that may aid in the development of new drugs. In this sense, the present study aims to demonstrate the mechanism of interaction for the activation of NAB and NAT molecules by cytochrome P450 2A13 [15,16].
In this paper, we have described the molecular docking results of NAB and NAT ligands interaction with cytochrome P450 2A13. We also performed Molecular Dynamics (MD) simulation to evaluate the intermolecular interactions during the period of the simulation. Finally, we studied the spontaneity of the formation of the systems and their energetic components by means of free energy calculations using the Molecular Mechanics/Generalized-Born Surface Area (MM/GBSA) methods.
MATERIALS AND METHODS
Molecular docking
Molecular docking studies were performed using the software Molegro Virtual Docker 5.5 (MVD 5.5) [20]. The crystallographic structure of cytochrome P450 2A13, used as a receptor, can be located in the Protein Data Bank (PDB) with ID: 4EJG [16]. The MolDock scoring function (GRID) was used with a grid resolution of 0.30 Å and a radius of 6 Å encompassing the entire bond cavity.
Molecular dynamics simulation
The protein inhibitor system was constructed for the simulation using the tleap modulus of AMBER 16 [25,26]. The General Amber Force Field (GAFF) [27] and ff14SB [28] force field was used for all the solvent-solvent simulations with the TIP3P [29] model water molecules. The system was solvated in a truncated octahedron periodic box. The cut-off radius was 12 Å for all solvent atoms relative to the solute in the x, y and z axes and a suitable number of counter-ions were added to the system to neutralize their charge.
Energy minimization was performed in the AMBER 16 sander module. At each stage, 5000 cycles were performed using the steepest descent method and conjugate gradient algorithm. In the first, the protein and inhibitor were fixed by applying a harmonic force constant of 200 kcalmol-1Å-2, the water molecules being free to move. In the following two, the restraint strength was reduced, so that in the final stage the entire system would be able to move freely.
After minimization, the systems were gradually heated from 0 to 300k in five steps, for a total time of 600ps. In four of these, a constant force harmonic of 25kcalmol-1Å-2 was used to restrict heavy atoms. In the final step, the restraining force was reduced to zero so that the whole solute was free to move. These stages were performed using the Langevin thermostat [30] with a collision frequency of 3.0 ps-1.
Following this, in order to obtain system equilibrium, a simulation of 2ns was performed at a temperature of 300k, with constant pressure without restriction. The calculation of the electrostatic interactions was performed using the Particle Mesh Ewald method [31] and the SHAKE [32] algorithm was utilized to restrict the bond lengths involving the hydrogen atoms. Afterward, molecular dynamics simulations of production up to 100ns were performed for all systems, unrestrained in the NVT ensemble.
Free energy calculation
ΔGbind = Gcomplex - Greceptor - Gligand (1)
ΔGbind = ΔH - TΔS = EMM + ΔGsolv – TΔS (2)
ΔEMM = ΔEinternal + ΔEelectrostatic + ΔEvdW (3)
ΔGsolv = ΔGGB + ΔGnonpol (4)
In which ΔGbind is the free energy of the protein-ligand binding, resulting from the sum of the molecular mechanics energy (ΔEMM), the desolvation free energy (ΔGsolv), and the entropy (-TΔS). The energy of molecular gas phase mechanics (ΔEMM) can be described by the sum of the internal energy contributions (ΔEinternal), the sum of the connection, angle and dihedral energies), electrostatic contributions (ΔEeletrostatic) and van der Waals terms (ΔEvdW). Desolvation free energy (ΔGsolv) is the sum of the polar (ΔGGB) and non-polar (ΔGnonpol) contributions. The polar desolvation term was calculated using the implicit Generalized Born (GB) approaches.
RESULTS AND DISCUSSION
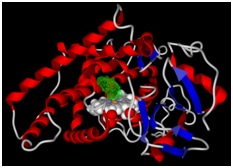
Figure 2: The binding pocket of the molecules is represented by green color.
This cytochrome was chosen because it is expressed in the respiratory tract [35] and is responsible for the metabolism of nitrosamines [36].The molecular docking simulation of these nitrosamines with cytochrome was performed because NAB [37] and NAT [38] are carcinogenic agents present in tobacco smoke. In several studies they have been reported as being responsible for causing lung cancer [39-41]. Therefore, elucidation of the binding mechanism of these molecules is important for the study of their metabolism. The molecular docking results are summarized in table 1.
|
Molecules |
MolDock Scorea |
Rerank Scoreb |
Interaction (kcal/mol)c |
Contribution of the Cofactord |
Internale |
LE1f |
LE3g |
|
NAB |
-86.74 |
-72.07 |
-97.96 |
-17.28 |
11.22 |
-6.19 |
-5.14 |
|
NAT |
-82.86 |
-71.82 |
-99.8 |
-11.03 |
16.93 |
-5.91 |
-5.12 |
The MolDock score found for NAB (-86.74) and NAT (-82.86) at the P450 binding site was favorable for the ligand receptor interaction, as it favored the molecular stability of the system. In addition, the affinity values for the binding of the molecules to the cytochrome were favorable, demonstrating that these compounds were capable of binding to cytochrome to perform productive interactions. For NAB and NAT, these values were -97,963 kcal/mol and -99,803 kcal/mol respectively. The energy contribution of the HEME prosthetic group was also calculated and its values were found favorable in both the systems. The energy contribution for the molecules NAB and NAT were -17.281 kcal/mol and -11.036 kcal/mol respectively.
With the above obtained results it became possible to verify the interactions between the molecules and the residues of the protein and the HEME prosthetic group.
These interactions are described in the following tables 2 and 3 and figure 3a and 3b.
|
Molecule |
Residue |
Group Heme |
Interaction |
Distance (Å) |
|
P30 |
Ala301-CB |
- |
Hidrophobic: Alkyl |
4.18 |
|
P31 |
Ala301-CB |
- |
Hidrophobic: Pi-Alkyl |
5.32 |
|
- |
Ala301-CB |
B |
Hidrophobic: Pi-Alkyl |
4.05 |
|
- |
Ala301-CB |
A |
Hidrophobic: Pi-Alkyl |
4.69 |
|
P30 |
- |
A |
Hidrophobic: Pi-Alkyl |
4.42 |
|
P30 |
- |
C |
Hidrophobic: Pi-Alkyl |
5.36 |
|
Molecule |
Residue |
Group Heme |
Interaction |
Distance (Å) |
|
O1 |
Ala301-CA |
- |
Hydrogen Bond |
3.47 |
|
P20 |
Ala301-CB |
- |
Hydrophobic: Alkyl |
3.78 |
|
P21 |
Ala301-CB |
- |
Hydrophobic: Pi-Alkyl |
5.21 |
|
- |
Ala301-CB |
A |
Hydrophobic: Pi-Alkyl |
4.69 |
|
- |
Ala301-CB |
B |
Hydrophobic: Pi-Alkyl |
4.05 |
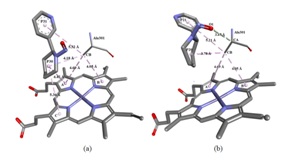
Figure 3: Molecular interactions. (a) Molecular interactions between NAB and CYP2A13 and HEME group. (b) Molecular interactions between NAT and CYP2A13 and HEME group.
The NAT molecule, having an interaction energy value of -99.803 kcal/mol, showed the best interaction with the active site of the protein in the docking simulation. This molecule showed three interactions with the alanine residue. The atom O1 of the molecule interacted with the CA atom of alanine 301 by means of hydrogen bonding at a distance of 3.47 Å. In this interaction the NAT molecule is the acceptor and the alanine molecule is the donor of the hydrogen atom. The CB atom interacted with the P21 ring by means of a pi-alkyl interaction at a distance of 5.21 Å while it interacted with the Pyridine ring (P20) through an alkyl interaction at a distance of 3.78 Å. The pyrrole rings A and B of the HEME group interacted with the alanine residue by means of hydrophobic pi-alkyl type interaction at the distances of 4.69 and 4.05 Å, respectively.
Analysis of the stability of the three-dimensional structure of protein and ligands
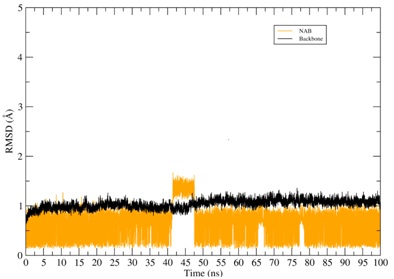
Figure 4: RMSD of the CYP2A13-ligands systems. The RMSD of the protein backbone is represented in black color while the RMSD of the binder is represented in various other colors.
Throughout the simulation of the NAT its structural conformation underwent changes alternating its state between moments of molecular balance and small conformational variations. The NAB molecule also maintained its equilibrium at the CYP site, undergoing small conformational changes during the simulation. Throughout the simulation time the NAB and NAT compounds remained bound to the CYP binding pocket.
Analysis of free energy and its components
|
Ligand |
ΔEvdw |
ΔEele |
ΔGele,sol |
ΔGnpol,sol |
ΔEnon-polar |
ΔEpolar |
ΔGbind |
|
NAB |
-34.97 ± 0.04 |
-9.50 ± 0.05 |
10.31 ± 0.02 |
-4.02 ± 0.01 |
-38.99 |
1.07 |
-38.19 ± 0.05 |
|
NAT |
-31.78 ± 0.04 |
-7.92 ± 0.05 |
12.62 ± 0.03 |
-4.01 ± 0.01 |
-35.79 |
4.70 |
-31.08 ± 0.05 |
According to the values obtained with the free energy calculations, the binders showed the following bond affinity values: CYP-NAB: -38.19 and CYP-NAT: -31.08.
In addition to the absolute values of free energy, its components were also calculated so that the mechanism of interaction of the molecules with the protein is better understood. In all the systems the van de Waals interactions (ΔEvdW) and the electrostatic interactions of the gas phase (ΔEele) favored the bond free energy (ΔGbind) of the complexes. The values for nonpolar solvation energies (ΔGnpol,sol) and the non-polar (ΔEnon-polar) contributions were similar to each other and also favored the ligand receptor interaction.
CONCLUSION
ACKNOWLEDGMENT
DISCLOSURE
AUTHOR’S CONTRIBUTIONS
REFERENCES
- US Department of Health and Human Services (2010) How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease. US Department of Health and Human Services, Washington D.C., USA.
- Pinheiro MDA, Torres LF, Bezerra MS, Cavalcante RC, Alencar RD, et al. (2017) Prevalence and associated factors of alcohol consumption and smoking among medical students in northeastern Brazil. Rev Bras Educ Med 41: 231-239.
- Ng M, Freeman MK, Fleming TD, Robinson M, Dwyer-Lindgren L, et al. (2014) Smoking prevalence and cigarette consumption in 187 countries, 1980-2012. JAMA 311: 183-192.
- Frieden TR, Jaffe HW, Richards CL, Iademarco MF, Cono J (2014) Current cigarette smoking among adults-United States, 2005-2012. Centers for Disease Control and Prevention, Morbidity and Mortality Weekly Report 63: 29-34.
- Pesch B, Kendzia B, Gustavsson P, Jöckel KH, Johnen G, et al. (2012) Cigarette smoking and lung cancer--relative risk estimates for the major histological types from a pooled analysis of case-control studies. Int J Cancer 131: 1210-1219.
- Barrera R, Shi W, Amar D, Thaler HT, Gabovich N, et al. (2005) Smoking and timing of cessation: Impact on pulmonary complications after thoracotomy. Chest 127: 1977-1983.
- Clifford GM, Lise M, Franceschi S, Egger M, Bouchardy C, et al. (2012) Lung cancer in the Swiss HIV cohort study: Role of smoking, immunodeficiency and pulmonary infection. Br J Cancer 106: 447-452.
- Mao Y, Yang D, He J, Krasna MJ (2016) Epidemiology of Lung Cancer. Surg Oncol Clin N Am 25: 439-445.
- Islami F, Torre LA, Jemal A (2015) Global trends of lung cancer mortality and smoking prevalence. Transl Lung Cancer Res 4: 327-338.
- Ahmad A, Gadgeel S (2016) Lung Cancer and Personalized Medicine, Springer International Publishing, Cham, Switzerland.
- Hoffmann D, Hecht SS (1985) Nicotine-derived N-nitrosamines and tobacco-related cancer: Current status and future directions. Cancer Res 45: 935-944.
- Ramírez N, Özel MZ, Lewis AC, Marcé RM, Borrull F, et al. (2014) Exposure to nitrosamines in thirdhand tobacco smoke increases cancer risk in non-smokers. Environ Int 71: 139-147.
- Wong HL, Zhang X, Zhang QY, Gu J, Ding X, et al. (2005) Metabolic activation of the tobacco carcinogen 4-(methylnitrosamino)-(3-pyridyl)-1-butanone by cytochrome P450 2A13 in human fetal nasal microsomes. Chem Res Toxicol 18: 913-918.
- Shimada T (2017) Inhibition of carcinogen-activating cytochrome P450 enzymes by xenobiotic chemicals in relation to antimutagenicity and anticarcinogenicity. Toxicol Res 33: 79-96.
- Schlicht KE, Berg JZ, Murphy SE (2009) Effect of CYP2A13 active site mutation N297A on metabolism of coumarin and tobacco-specific nitrosamines. Drug Metab Dispos 37: 665-671.
- DeVore NM, Scott EE (2012) Nicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone binding and access channel in human cytochrome P450 2A6 and 2A13 enzymes. J Biol Chem 287: 26576-26585.
- Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98: 5648-5652.
- Lee C, Yang W, Parr RG (1988) Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys Rev B Condens Matter 37: 785-789.
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, et al. (2009) Electronic Supplementary Material (ESI) for Chemical Science. The Royal Society of Chemistry 1-20.
- Thomsen R, Christensen MH (2006) MolDock: A new technique for high-accuracy molecular docking. J Med Chem 49: 3315-3321.
- Bayly CI, Cieplak P, Cornell W, Kollman PA (1993) A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J Phys Chem 97: 10269-10280.
- Cornell WD, Cieplak P, Bayly CI, Kollman PA (1993) Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of solvation. J Am Chem Soc 115: 9620-9631.
- Wang J, Cieplak P, Kollman PA (2000) How well does a Restrained Electrostatic Potential (RESP) model perform in calculating conformational energies of organic and biological molecules? Journal of Computational Chemistry 21: 1049-1074.
- Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA (2004) PDB2PQR: An automated pipeline for the setup of poisson-boltzmann electrostatics calculations. Nucleic Acids Res 32: 665-667.
- Case DA, Cheatham TE 3rd, Darden T, Gohlke H, Luo R, et al. (2005) The Amber biomolecular simulation programs. J Comput Chem 26: 1668-1688.
- Cruz JN, Costa JFS, Khayat AS, Kuca K, Barros CAL, et al. (2018) Molecular dynamics simulation and binding free energy studies of novel leads belonging to the benzofuran class inhibitors of mycobacterium tuberculosis polyketide synthase 13. J Biomol Struct Dyn 4: 1-12.
- Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) Development and testing of a general amber force field. J Comput Chem 25: 1157-1174.
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, et al. (2015) ff14sb: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 11: 3696-3713.
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics 79: 926.
- Izaguirre JA, Catarello DP, Wozniak JM, Skeel RD (2001) Langevin stabilization of molecular dynamics. J Chem Phys 114: 2090.
- Darden T, York D, Pedersen L (1998) Particle mesh Ewald: An N•log(N) method for ewald sums in large systems. J Chem Phys 98: 10089.
- Ryckaert JP, Ciccotti G, Berendsen HJC (1977) Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J Comput Phys 23: 327-341.
- Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, et al. (2000) Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc Chem Res 33: 889-897.
- HouJ T, Wang Y, Li Y, Wang W (2011) Assessing the performance of the MM/PBSA and MM/GBSA methods.1.The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model 51: 69-82.
- Chiang HC, Wang CK, Tsou TC (2012) Differential distribution of CYP2A6 and CYP2A13 in the human respiratory tract. Respiration 84: 319-326.
- Jalas JR, Hecht SS, Murphy SE (2005) Cytochrome P450 enzymes as catalysts of metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco specific carcinogen. Chem Res Toxicol 18: 95-110.
- Hecht SS, Stepanov I, Carmella SG (2016) Exposure and Metabolic Activation Biomarkers of Carcinogenic Tobacco-Specific Nitrosamines. Acc Chem Res 49: 106-114.
- Xue J, Yang S, Seng S (2014) Mechanisms of Cancer Induction by Tobacco-Specific NNK and NNN. Cancers (Basel) 6: 1138-1156.
- Wong HL, Murphy SE, Hecht SS (2005) Cytochrome P450 2A-catalyzed metabolic activation of structurally similar carcinogenic nitrosamines: N'-nitrosonornicotine enantiomers, N-nitrosopiperidine, and N-nitrosopyrrolidine. Chem Res Toxicol 18: 61-69.
- Hecht SS (1998) Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol 11: 559-603.
- Carlson ES, Upadhyaya P, Hecht SS (2016) Evaluation of nitrosamide formation in the cytochrome P450-mediated metabolism of tobacco-specific nitrosamines. Chem Res Toxicol 29: 2194-2205.
Citation: Cruz JN, Oliveira MS, Vogado JH, Silva SG, Costa WA, et al., (2018) Molecular Insights on the Interactions of Nitrosamines from Cigarette Smoking with Cyp2a13 using Molecular Docking and Molecular Dynamics Simulation. J Pulm Med Respir Res 4: 018.
Copyright: © 2018 Jorddy N Cruz, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.