Mepolizumab for Eosinophilic Granulomatous Polyangiitis (Churg-Strauss Syndrome)
*Corresponding Author(s):
John S PixleyDepartment Of Internal Medicine, Professor And Chief Rheumatology/Immunology Division, Texas Tech University Health Science Centre, 3601 4th Street MS 9410 Lubbock, Texas 79430, United States
Tel:806 743-1683,
Email:John.Pixley@ttuhsc.edu
SHORT COMMENTARY
Eosinophilic granulomatous polyangiitis (EGPA) is a rare systemic vasculitis characterized by asthma, sinus disease and varied extra-respiratory tract vasculitic manifestations (Table 1) [1]. It is likely a variant of two other anti-neutrophil cytoplasmic antibody (ANCA-associated-vasculitides or AAV): granulomatous polyangiitis (GPA) and microscopic polyangiitis (MPA). This is due to the finding of ANCA positivity in some patients and overlap symptom manifestations of small vessel vasculitis such as neuropathy, glomerulonephritis and palpable purpura. While it has been proposed that ANCAs are pathogenic, a significant minority of patients with granulomatous polyangiitis (GPA), microscopic polyangiitis (MPA) and ~ 50-60% of EGPA patients do not express ANCAs in the circulation [2,3]. The absence of ANCA in ~ 50% of patients with EGPA has fostered speculation that there is a primarily asthmatic form termed “hypereosinophilia with (any) systemic (non-vasculitic) manifestations” (HASM) in comparison to a second form with overt polyangiitis. Problematic to this proposal is the observation that ANCA negative patients with EGPA are also found to have vasculitic manifestations. An alternative explanation suggests that the overt vasculitic manifestations represent more advanced disease [4,5].
Table 1: Main clinical characteristics at diagnosis of the 383 patients with EGPA, according to ANCA status* [1].
|
Characteristic |
All (n=383) |
ANCA status unknown (n=35) |
ANCA-positive patients (n=108) |
ANCA-negative patients (n=240) |
P? |
|
Sex, no. male/female |
199/184 |
14/21 |
60/48 |
125/115 |
0.55 |
|
Age at diagnosis, mean ± SD years |
50.3 ± 15.7 |
47.5 ± 15.7 |
52.5 ± 15.3 |
49.6 ± 15.8 |
0.15 |
|
Diagnosis in 1996 or before, no. (%) |
128 (33.4) |
33 (94.3 |
27 (25.0) |
68 (28.3) |
0.52 |
|
History of allergy, no. (%) |
104 (27.2) |
9 (25.7) |
31 (28.7) |
64 (26.7) |
0.69 |
|
Asthama, no. (%) |
349 (91.1) |
31 (88.6) |
100 (92.6) |
218 (90.8) |
0.59 |
|
Duration of asthama at EGPA diagnosis, mean ± SD years |
9.3 ± 10.8 |
7.6 ± 14.2 |
10.0 ± 11.7 |
9.4 ± 9.7 |
0.77 |
|
Symptoms and manifestaions, no. (%) |
|
|
|
|
|
|
Weight loss |
189 (49.3) |
25 (71.4) |
62 (57.4) |
102 (42.5) |
0.01 |
|
Mean ± SD kg lost |
7.8 ± 4.7 |
10.6 ± 7.0 |
7.8 ± 4.4 |
7.3 ± 4.1 |
0.44 |
|
Fever |
149 (38.9) |
22 (62.9) |
44 (40.7) |
83 (34.6) |
0.27 |
|
Myalgias |
149 (38.9) |
14 (40.0) |
44 (40.7) |
91 (37.9) |
0.62 |
|
Arthralgias |
114 (29.8) |
13 (37.1) |
37 (34.3) |
64 (26.7) |
0.15 |
|
ENT manifestaions |
184 (48.0) |
14 (40.0) |
64 (59.3) |
106 (44.2) |
<0.01 |
|
Rhinitis |
65 (17.0) |
4 (11.4) |
27 (25.0) |
34 (14.2) |
0.01 |
|
Sinusitis/polyposis |
160 (41.8) |
13 (37.1) |
56 (51.9) |
91 (37.9) |
0.02 |
|
Lung manifestaions |
350 (91.4) |
31 (88.6) |
100 (92.6) |
219 (91.3) |
0.68 |
|
Chest pain |
28 (7.3) |
0 |
6 (5.6) |
22 (9.2) |
0.25 |
|
Lung nodule(s) |
20 (5.2) |
1 (2.9) |
9 (8.3) |
10 (4.2) |
0.11 |
|
Lung infiltartates |
148 (38.6) |
10 (28.6) |
44 (40.7) |
94 (39.2) |
0.78 |
|
Pleural effusion |
34 (8.9) |
1 (2.9) |
5 (4.6) |
28 (11.7) |
0.04 |
|
Alveolar hemorrhage |
16 (4.2) |
1 (2.9) |
8 (7.4) |
7 (2.9) |
0.06 |
|
Cutaneous manifestations |
152 (39.7) |
16 (45.7) |
49 (45.4) |
87 (36.3) |
0.11 |
|
Purpura |
86 (22.5) |
8 (22.9) |
31 (28.7) |
47 (19.6) |
0.06 |
|
Pseudo-urticarial rash, hives |
38 (9.9) |
1 (2.9) |
13 (12.0) |
24 (10.0) |
0.57 |
|
Subcutaneous nodule (s) |
37 (9.7) |
8 (22.9) |
9 (8.3) |
20 (8.3) |
0.99 |
|
Livedo reticularis |
15 (3.9) |
3 (8.6) |
2 (1.9) |
10 (4.2) |
0.27 |
|
Gangrenous necrotic lesions |
14 (3.7) |
0 |
5 (4.6) |
9 (3.8) |
0.7 |
|
Neurologic symptoms |
211 (55.1) |
25 (71.4) |
72 (66.7) |
115 (47.5) |
<0.01 |
|
Peripheral neuropathy |
197 (51.4) |
23 (65.7) |
68 (63.0) |
106 (44.2) |
<0.01 |
|
Mononeuritis multiplex |
176 (46.0) |
23 (65.7) |
59 (54.6) |
94 (39.2) |
<0.01 |
|
CNS involvement |
20 (5.2) |
4 (11.4) |
7 (6.5) |
9 (3.8) |
0.26 |
|
Cranial nerve involvement |
12 (3.1) |
3 (8.6) |
3 (2.8) |
6 (2.5) |
0.88 |
|
Cardiovascular manifestations |
105 (27.4) |
12 (34.3) |
20 (18.5) |
73 (30.4) |
0.02 |
|
Raynaud's phenomenon |
6 (1.6) |
0 |
2 (1.9) |
4 (1.7) |
0.9 |
|
Cardiomyopathy (FFS item) |
63 (16.4) |
8 (22.9) |
9 (8.3) |
46 (19.2) |
0.01 |
|
Pericarditis |
58 (15.1) |
4 (11.4) |
14 (13.0) |
40 (16.7) |
0.38 |
|
Deep venous thrombosis/pulmonary embolism |
29 (7.6) |
1 (2.9) |
8 (7.4) |
20 (8.3) |
0.77 |
|
Gastrointestinal involvement |
89 (23.2) |
9 (25.7) |
24 (22.2) |
56 (23.3) |
0.82 |
|
Abdominal pain |
78 (20.4) |
6 (17.1) |
21 (19.4) |
51 (21.3) |
0.7 |
|
Surgical abdomen |
22 (5.7) |
1 (2.9) |
3 (2.8) |
18 (7.5) |
0.09 |
|
FTS-defined manifestations |
7 (1.8) |
0 |
1 (0.9) |
6 (2.5) |
0.33 |
|
Eye involvement |
25 (6.5) |
2 (5.7) |
9 (8.3) |
14 (5.8) |
0.39 |
|
Renal manifestations |
83 (21.7) |
15 (42.9) |
29 (26.9) |
39 (16.3) |
0.02 |
|
Proteinurea>0.4gm/24hours |
49 (12.8) |
6 (17.1) |
24 (22.2) |
19 (7.9) |
<0.01 |
|
Creatinine>140 µmoles/liter (FFS item) ? |
11 (4.3) |
1 (4.3) |
5 (6.2) |
5 (3.3) |
0.31 |
|
BVAS at diagnosis, mean ± SD |
19.1 ± 8.4 |
18.1 ± 6.2 |
20.7 ± 8.6 |
18.4 ± 8.6 |
0.05 |
|
FFS, no. (0%) |
|
|
|
|
|
|
0 |
291 (76.0) |
23 (65.7) |
87 (80.6) |
181 (75.4) |
0.59 |
|
1 |
82 (21.4) |
11 (31.4) |
18 (16.7) |
53 (22.1) |
|
|
≥2 |
10 (2.6) |
1 (2.9) |
3 (2.8) |
6 (2.5) |
|
|
Revised FFS, no. (%) |
|
|
|
|
|
|
0 |
98 (25.6) |
10 (28.6) |
35 (32.4) |
53 (22.1) |
0.1 |
|
1 |
120 (31.3) |
6 (17.1) |
37 (34.3) |
77 (32.1) |
|
|
≥2 |
165 (43.1) |
19 (54.3) |
36 (33.3) |
110 (45.8) |
|
*EGPA= eosinophilic granulomatosis with polyangiitis (Churg-Strauss); ENT = ear, nose, throat; CNS = central nervous system; FFS = Five-Factors Score; BVAS = Brimingham Vasculitis Activity Score.
?For comparison of antineutrophil cytoplasmic antibody (ANCA)-positive patients versus ANCA-negative patients.
?Serum creatinine levels at diagnosis were available for 254 patients (23 patients with unknown ANCA-positive patients, and 150 ANCA-negative patients).
EGPA is classically considered a T helper-2 (Th-2) mediated disease based on analysis of peripheral blood, bronchial alveolar lavage fluid and tissue samples from affected patients [6,7]. Besides Th-2, Th-1 and Th-17 differentiation pathways are also thought to be operative in EGPA. Isolated T cells in each of these pathways express different cytokine profiles (Figure 1). B cell activation may also play a role based in part on elevations in circulating IgE in affected patients. Finally, the observation of increased levels of interleukin 25 (IL25) in the circulation suggests eosinophils themselves may augment TH2 polarity [8].
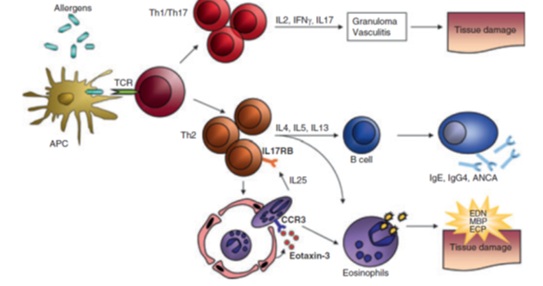 Figure 1: Simplified scheme of pathophysiological events in eosinophilic granulomatosis with polyangiitis (EGPA) (Churg-Strauss). Hitherto unidentified allergens elict an adaptive immune response in EGPA patients. T cells secrete Th1- (IFN-), Th17- (IL-17) and Th2- (IL-4, IL-3, IL-5) associated cytokines and activate eosinophils. The strong Th2 immune response precipitates a B-cell response resulting in IgG4, IgE and antineutrophil cytoplasmic antibodies (ANCA) production. Increased expression an secretion of eotaxin-3 guides eosinphils to the endithelium and tissues. Eosinophils in turn maintain a vicious circle of T-cell activation by secreting IL-25. Local degranulation of activated eosinphils finally causes damage, necrosis and fibrosis to tissues an vessels. APC, antigen-presenting cell; TCR. T-cell receptor; ANCA, anti-neutrophil cytoplasmic antibody; EDN, eosinophil-derived neurotosin; MBP, major basic protein; ECP, eosinophilic cationic protein [6].
Figure 1: Simplified scheme of pathophysiological events in eosinophilic granulomatosis with polyangiitis (EGPA) (Churg-Strauss). Hitherto unidentified allergens elict an adaptive immune response in EGPA patients. T cells secrete Th1- (IFN-), Th17- (IL-17) and Th2- (IL-4, IL-3, IL-5) associated cytokines and activate eosinophils. The strong Th2 immune response precipitates a B-cell response resulting in IgG4, IgE and antineutrophil cytoplasmic antibodies (ANCA) production. Increased expression an secretion of eotaxin-3 guides eosinphils to the endithelium and tissues. Eosinophils in turn maintain a vicious circle of T-cell activation by secreting IL-25. Local degranulation of activated eosinphils finally causes damage, necrosis and fibrosis to tissues an vessels. APC, antigen-presenting cell; TCR. T-cell receptor; ANCA, anti-neutrophil cytoplasmic antibody; EDN, eosinophil-derived neurotosin; MBP, major basic protein; ECP, eosinophilic cationic protein [6].
Based on overlap between the other 2 forms of AAV treatment has centered on the use of systemic steroids and alkylating agents for patients with severe disease. A five factor score to gauge severity has been proposed to support the addition of cyclophosphamide to systemic steroids [9]. Current consensus and clinical experience support the effectiveness of cyclophosphamide for this condition. In addition, the well-recognized complications from the use long-term high dose steroid therapy requires the addition non-alkylating immunosuppressants and/or rituximab to minimize steroid side effects. Thus, immunosuppressive agents (methotrexate or azathioprine) are commonly added either to maintain remission or as steroid sparing agents [10]. The level of evidence in support of the use of these agents is 1B based on up to Date’s analysis [11]. Clinical experience has supported this view. Unfortunately, it is difficult to organize and finance investigation of treatment outcome for a rare disease such as EGPA when there is an absence of industry support. Nevertheless, support for this treatment protocol is supported by marked improvement in outcome as EGPA was universally fatal prior to the use of glucocorticoids [1,6].
The discovery of increased levels of IL5 in the airways of patients with EGPA and its effectiveness in eosinophilic asthma prompted an investigator initiated study demonstrating modest benefit with the use of mepolizumab (an IL5 inhibitor) in patients with moderate EGPA. This proof of concept study in patients with remitting/refractory disease on varying doses of prednisone
What to make of this study! While mepolizumab offers a degree of disease amelioration, it supports the concept that multiple arms of T helper cell differentiation are the primary culprit in EGPA with eosinophilic activation a terminal factor in immune pathology. Thus immunosuppressive agents are required to dampen T cell activation (Figure 1). In the active treatment arm 22% had a major relapse and 47% did not achieve remission despite continuing immunosuppressive medications in 60% of patients. Unfortunately, the data presented in the report and appendix does not identify remission incidence (prednisone/prednisolone dose <= 4mg/day) in patients on concomitant immunosuppressives. By comparison, a retrospective look at rituximab therapy in 41 patients (44% on concomitant immunosuppressives) found 88% reached complete (49%) or partial (39%) remission with comparable reduction in steroid dosing [14].
Rheumatologists commonly treat relatively rare diseases associated with inadequate clinical trial data to guide therapy. Despite these limitations retrospective analyses support the addition of immunosuppressive therapy to aid in lower steroid dosing and subsequent steroid side effects. These studies and their use in other serious rheumatic diseases support their relative safety. The question posed regarding mepolizumab use in EGPA remains open in comparison to rituximab. Further study is required to answer this question.
REFERENCES
- Comarmond C, Pagnoux C, Khellaf M, Cordier JF, Hamidou M, et al. (2013) French Vasculitis Study Group. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): Clinical characteristics and long- term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum 65: 270-281.
- https://onlinelibrary.wiley.com/doi/full/10.1002/art.37721
- Radice A, Bianchi L, Sinico RA (2013) Anti-neutrophil cytoplasmic autoantibodies: Methodological aspects and clinical significance in systemic vasculitis. Autoimmun Rev 12: 487-495.
- Cottin V, Bel E, Bottero P, Dalhoff K, Humbert M, et al. (2017) Revisiting the systemic vasculitis in eosinophilic granulomatosis with polyangiitis (Churg-Strauss): A study of 157 patients by the Groupe d'Etudes et de Recherche sur les Maladies Orphelines Pulmonaires and the European Respiratory Society Taskforce on eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Autoimmun Rev 16: 1-9.
- Greco A, Rizzo MI, De Virgilio A, Gallo A, Fusconi M, et al. (2014) Churg-Strauss syndrome. Autoimmun Rev 14: 341-348.
- Vaglio A, Buzio C, Zwerina J (2013) Eosinophilic granulomatosis with polyangiitis(Churg-Strauss): State of the art. Allergy 68: 261-273.
- Jakiela B, Szczeklik W, Plutecka H, Sokolowska B, Mastalerz L, et al. (2012) Increased production of IL-5 and dominant Th2-type response in airways of Churg-Strauss syndrome patients. Rheumatology (Oxford) 51: 1887-1893.
- Terrier B, Bièche I, Maisonobe T, Laurendeau I, Rosenzwajg M, et al. (2010) Interleukin-25: A cytokine linking eosinophils and adaptive immunity in Churg-Strauss syndrome. Blood 116: 4523-4531.
- Guillevin L, Pagnoux C, Seror R, Mahr A, Mouthon L, et al. (2011) The Five-Factor Score revisited: Assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore) 90: 19-27.
- Groh M, Pagnoux C, Baldini C, Bel E, Bottero P, et al. (2015) Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) (EGPA) Consensus Task Force recommendations for evaluation and management. Eur J Intern Med 26: 545-553.
- https://www.uptodate.com/contents/treatment-and-prognosis-of-eosinophilic-granulomatosis-with-polyangiitis-churg-strauss
- Wechsler ME, Akuthota P, Jayne D, Khoury P, Kilon A (2017) Mepolizumab or Placebo for Eosinophilic; Granulomatosis with Polyangiitis. New England Journal of Medicine 376: 1921-1932.
- Kim S, Marigowda G, Oren E, Israel E, Wechsler ME (2010) Mepolizumab as a steroid-sparing treatment option in patients with Churg-Strauss syndrome. J Allergy Clin Immunol 125: 1336-1343.
- Mohammad AJ, Hot A, Arndt F, Moosig F, Guerry MJ, et al. (2016) Rituximab for the treatment of eosinophilic granulomatosis with polyangiitis (Churg-Strauss). Ann Rheum Dis 75: 396-401.
Citation: Pixley JS (2020) Mepolizumab for Eosinophilic Granulomatous Polyangiitis (Churg-Strauss Syndrome). J Clin Immunol Immunother 6: 015.
Copyright: © 2020 John S Pixley, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.