The Interoceptive Antireward Pathway and Gut Dysbiosis in Addiction
*Corresponding Author(s):
Sean J. O’SullivanDaniel Baugh Institute For Functional Genomics And Computational Biology, Department Of Pathology, Anatomy And Cell Biology, Thomas Jefferson University, Philadelphia, PA, United States/ Brain Stimulation Lab, Stanford University, Stanford, CA, United States
Tel:+1 610-547-5327,
Email:sjo003@stanford.edu
Mini Review
The concept of antireward as a motivator for drug addiction was first proposed in 2005 by Koob and Le Moal [1]. This model conjectures that addictive behaviors, most often the use of substances of abuse, have two positively reinforcing motivators: the stimulation of reward and the inhibition of antireward (Figure1) [2]. Furthermore, opponent-process functionality inherent in biological systems predicts an absence of reward and a stimulation of antireward in absence of the addictive behavior; or abstinence from the xenobiotic in the case of substance dependence (ie withdrawal) [3]. That is, there are also two negative reinforcement motivators for drug-seeking and taking. Biological evidence for a reward pathway in addiction is well-established. Mesolimbic dopamine is stimulated by substance use leading to the activation of pleasure centers, and this pathway is, in fact, depressed in substance abstinence leading to anhedonia [4,5]. The biological evidence for an antireward pathway has been accumulating since Koob and Le Moal’s original article suggesting signaling in the extended amygdale.
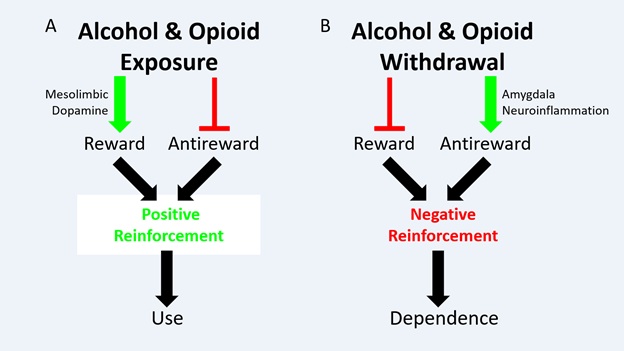 Figure 1: Schematic of Opponent-Process Model in Addiction. (A) Alcohol and/or opioid exposure has two effects: Stimulate reward, via the mesolimbic dopamine pathway, and inhibit antireward. These effects motivate substance use via positive reinforcement (B) Alcohol and/or opioid withdrawal has two effects. Inhibit reward, by inhibiting the mesolimbic dopamine pathway (not shown), and stimulate antireward. This review proposes that amygdala neuroinflammation mediated primarily by astrocytes is an endpoint in antireward stimulation in alcohol and opioid withdrawal, though this hypothesis warrants further testing. These effects, whatever the mechanism, motivate substance dependence via negative reinforcement. Originally published in O’Sullivan et al. 2021.
Figure 1: Schematic of Opponent-Process Model in Addiction. (A) Alcohol and/or opioid exposure has two effects: Stimulate reward, via the mesolimbic dopamine pathway, and inhibit antireward. These effects motivate substance use via positive reinforcement (B) Alcohol and/or opioid withdrawal has two effects. Inhibit reward, by inhibiting the mesolimbic dopamine pathway (not shown), and stimulate antireward. This review proposes that amygdala neuroinflammation mediated primarily by astrocytes is an endpoint in antireward stimulation in alcohol and opioid withdrawal, though this hypothesis warrants further testing. These effects, whatever the mechanism, motivate substance dependence via negative reinforcement. Originally published in O’Sullivan et al. 2021.
They to Koob and Le Moal proposed corticotrophin-releasing factor (CRF) signaling, in addition to norepinephrine (NE) and dynorphin, in the amygdale as substrates of antireward. The extended nucleus of the amygdala is an ostensible location to generate antireward motivations because of its proven function in threat detection, fear, and negative emotion [6]. Additionally, CRF as a substrate is compelling because CRF increases in the amygdala during withdrawal from alcohol [7], cocaine [8], nicotine [9], and cannabinoids [10]. Moreover, CRF antagonism in the amygdala decreases the anxiety-like behavior characteristic of alcohol and opioid withdrawal and also substance intake [11-15].
Our group has built on this work. We recently proposed a modified antireward model suggesting that antireward signaling in the amygdala, in both alcohol and opioid withdrawal, involves neuro inflammation mediated by astrocytes [2,16]. We further suggest that gut dysbiosis may contribute to this phenomenon via vagal afferents, gut peptide hormones, or additional blood-borne signaling (Figure 2). This hypothesis is supported by recent studies out of our lab suggesting neuro inflammation in the Central Nucleus of the Amygdala (CeA) and nucleus tractus solitarius (NTS) in opioid and alcohol withdrawal, respectively [16,17]. Additionally, gut dysbiosis was observed 24 hours following acute naltrexone-precipitated opioid withdrawal (Figure 3). These findings in context with other studies demonstrating gut dysbiosis in alcohol and opioid use and withdrawal [18], the effect of astrocyte-mediated neuro inflammation in the amygdala on anxiety-like behavior [19], and the emerging role of neuro inflammation in drug addiction and treatment [20-25], led us to a novel and proposal: The interceptive circuit connecting peripheral organs, especially the gut, to the CeA via the NTS contributes to antireward in both alcohol and opioid withdrawal by inciting neuro inflammation.
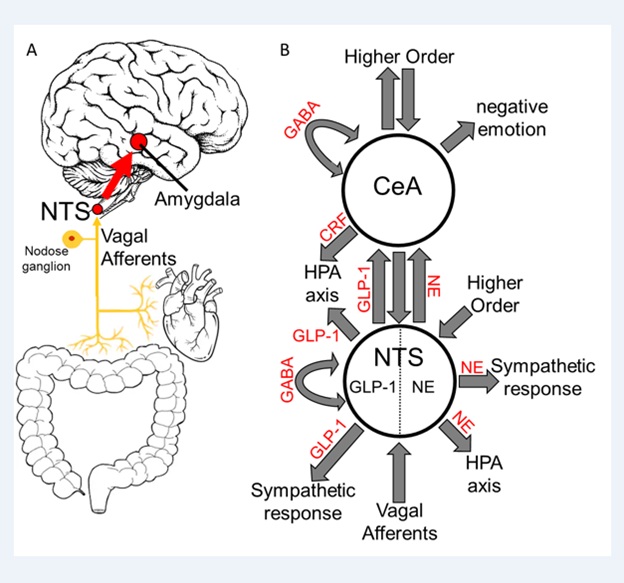 Figure 2: Interoceptive Vagal Circuit and Visceral-Emotional Neuraxis. (A) Interoceptive vagal afferents relay the state of the gut, which is highly influenced by gut microflora, and other peripheral organs to the Nucleus Tractus Solitarius (NTS). This information is subsequently relayed to the amygdala and influences emotional states.(B)A simplified cartoon representation showing the integrative roles of the Nucleus Tractus Solitarius (NTS) and the Central Nucleus of the Amygdala (CeA) in emotion, stress, and autonomic regulation. Two neuronal subtypes, GLP-1 and NE neurons, are highlighted. Many anatomical and functional connections are omitted for clarity (NE, Norepinephrine; GLP-1, Glucagon-Like Peptide 1; GABA, γ-Aminobutyric Acid; HPA axis, Hypothalamic-Pituitary-Adrenal axis; CRF, Corticotropin Releasing Hormone).Originally published in O’Sullivan et al. 2021.
Figure 2: Interoceptive Vagal Circuit and Visceral-Emotional Neuraxis. (A) Interoceptive vagal afferents relay the state of the gut, which is highly influenced by gut microflora, and other peripheral organs to the Nucleus Tractus Solitarius (NTS). This information is subsequently relayed to the amygdala and influences emotional states.(B)A simplified cartoon representation showing the integrative roles of the Nucleus Tractus Solitarius (NTS) and the Central Nucleus of the Amygdala (CeA) in emotion, stress, and autonomic regulation. Two neuronal subtypes, GLP-1 and NE neurons, are highlighted. Many anatomical and functional connections are omitted for clarity (NE, Norepinephrine; GLP-1, Glucagon-Like Peptide 1; GABA, γ-Aminobutyric Acid; HPA axis, Hypothalamic-Pituitary-Adrenal axis; CRF, Corticotropin Releasing Hormone).Originally published in O’Sullivan et al. 2021.
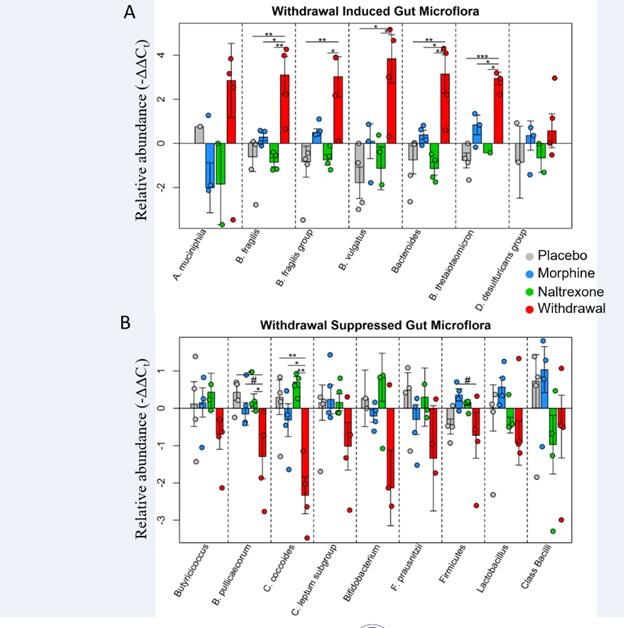 Figure 3: Relative abundance of gut microflora from rat cohort 2. Barplots display relative abundance of bacterial species (-ΔΔCt values). #p<0.1, *p<0.05, **p<0.008, ***p=0.0009; two-way ANOVA n=4 animals for each treatment. Originally published in O’Sullivan et al. 2019.
Figure 3: Relative abundance of gut microflora from rat cohort 2. Barplots display relative abundance of bacterial species (-ΔΔCt values). #p<0.1, *p<0.05, **p<0.008, ***p=0.0009; two-way ANOVA n=4 animals for each treatment. Originally published in O’Sullivan et al. 2019.
Others have speculated on the role of gut dysbiosis in substance dependence [18,26,27]. The importance of the gut-brain connection in health and disease and how gut microflora influence this connection is a growing area of investigation [28]. We have built on these advances by connecting this work to the antireward model of addiction. The neuro anatomy of the proposed interoceptive antireward pathway suggests that vagal afferents responding to gut dysbiosis transmit this information via synapses in the NTS which has strong bidirectional connections to the CeA (Figure 2) [29,30]. We conjecture this circuit influences CeA activity by glial-neuronal paracrine signaling or cytokines and chemokines, in addition to transmitters such as CRF, NE, and glucagon-peptide-1 (GLP-1).
Our study shows that astrocytes have the most perturbed transcriptome in the CeA 24 hours following acute naltrexone-precipitated opioid withdrawal [16]. This finding in astrocytes is consistent with the work of others that has yet to be published. The substrate in our dataset that emerged most prominently, however, was Tumor necrosis factor alpha (TNF-α). We found the transcript of this gene to be significantly up regulated in the withdrawal conditionin all three cell types collected-neurons, microglia, and astrocytes. TNF-α up regulation was confirmed with Western blot and immune fluorescence. We speculate that the signal for this elevation of TNF-α may originate in the gut where opioid receptors are numerous. And, that this neuro inflammation lowers the threshold of firing of neurons in the CeA resulting in hyperactivity that contributes to the negative emotion that characterizes substance withdrawal; That is, antireward stimulation [31,32].
This hypothesis is further supported by functional magnetic resonance imaging (fMRI) studies. Increased activity in the amygdala is observed during opioid craving in recently detoxed opioid-dependent patients [33]. Activity in the amygdala also increased upon anticipation of opioid use and subsequently decreased upon methadone administration [34]. Similarly, fMRI studies found that alcohol inhibited amygdala activity in heavy drinkers while hyperactive amygdala-orbitofrontal circuits in adolescents predicted future alcohol abuse [35,36]. These imaging studies suggest that, like the mesolimbic reward pathway, amygdale antireward functionality is involved in dependence of multiple substances. The contribution of gut dysbiosis via vagal afferents or other routes to these observations is not yet well understood. However, clinical treatments that target this pathway are likely to demonstrate efficacy for multiple substances of abuse.
One emerging treatment is the anti-neuro inflammatory ibudilast. This small molecule has multiple mechanisms of action including as a phosphodiesterase inhibitor and macrophage migration inhibitory factor (Mif) inhibitor [37]. In clinical trials, ibudilast has demonstrated efficacy in decreasing opioid cravings and withdrawal symptoms while increasing the analgesic effects of opioids in patients with dependence [23-25]. Additionally, ibudilast demonstrated efficacy in reducing reward in methamphetamine infusions [22,38]. This effect may have been due to reductions in peripheral inflammation implicating the interoceptive antireward pathway in ibudilast’s efficacy [39]. Ibudilast is currently in clinical trials for alcohol dependence as well [21]. The ability of this anti-inflammatory molecule to treat multiple forms of substance dependence is consistent with the antireward interoceptive pathway model.
The emergence of peripheral nerve stimulation for the treatment of opioid withdrawal adds further evidence to this model. The Food & Drug Administration (FDA) recently approved transcutaneous auricular neuro stimulation with the BRIDGE device to treat opioid withdrawal symptoms [40]. The proposed mechanism of action of this device is that stimulation of peripheral cranial nerves, which input into the NTS, subsequently stimulates the amygdala where “extracellular recordings from single cells in the rat amygdala before and during neuro stimulation with the BRIDGE device showed a 65% reduction in the baseline firing of neurons in the central nucleus of the amygdala” [41]. The effectiveness of the BRIDGE device in treating acute opioid withdrawal symptoms demonstrates that peripheral nerves influence an antireward center whose primary endpoint is the CeA. And, that targeting this pathway has potent effects on decreasing the physical and emotional pain that occurs in opioid withdrawal which may decrease drug-seeking and taking by decreasing negative reinforcement (Figure 1).
The evidence linking gut microflora dysbiosis to antireward, however, remains minimal. Studies have shown the importance of gut microflora in behavior and disease broadly, but their stimulation of antireward in acute withdrawal and protracted substance abstinence remains hypothetical [28,42]. Recent studies employing sub diaphragmatic vagotomy suggest vagal afferents do influence self-administration behavior [43], but the direct effects of microbes on this circuit and their sequalae influencing antireward in the amygdala remains an unknown frontier for further investigation.
Funding
This work was support by NIH HLB U01 HL133360 awarded to James S. Schwaber, Ph. D. and Rajanikanth Vadigepalli, Ph. D. and T32 AA-007463 awarded to Jan B. Hoek, Ph. D.
References
- Koob GF, Le Moal M (2005) Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nature Neuroscience 8: 1442-1444.
- O’Sullivan SJ, Schwaber JS (2021) Similarities in alcohol and opioid withdrawal syndromes suggest common negative reinforcement mechanisms involving the interoceptive antireward pathway. Neurosci. Biobehav. Rev 125: 355-364.
- Solomon RL, Corbit JD (1974) An opponent-process theory of motivation. I. Temporal dynamics of affect. Psychol. Rev 81: 119-45.
- Pierce RC, Kumaresan V (2006) The mesolimbic dopamine system: The final common pathway for the reinforcing effect of drugs of abuse? Neurosci. Biobehav. Rev 30: 215-238.
- Wise RA (2008) Dopamine and reward: The anhedonia hypothesis 30 years on. Neurotox. Res.14: 169-183.
- LeDoux J (2003) The emotional brain, fear, and the amygdala. Cell. Mol. Neurobiol 23: 727-738.
- Merlo-Pich E, Lorang M, Yeganeh M, Raber J, Koob GF, et al. (1995) Increase of extracellular corticotropin-releasing factor-like immune reactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J. Neurosci 15: 5439-5447.
- Richter RM, Weiss F (1999) In vivo CRF release in rat amygdala is increased during cocaine withdrawal in self-administering rats. Synapse 32: 254-261.
- George O, Ghozland S, Azar MR, Cottone P, Zorrilla EP, et al. (2007) CRF-CRF1 system activation mediates withdrawal-induced increases in nicotine self-administration in nicotine-dependent rats. Proc. Natl. Acad. Sci USA 104: 17198-17203.
- Rodríguez De, Fonseca F, Carrera MRA, Navarro M, Koob G, et al. (1997) Activation of corticotropin-releasing factor in the limbic system during cannabinoid withdrawal. Science 276: 2050-2054.
- Rassnick S, Heinrichs SC, Britton KT, Koob GF (1993) Microinjection of a corticotropin-releasing factor antagonist into the central nucleus of the amygdala reverses anxiogenic-like effects of ethanol withdrawal. Brain Res 605: 25-32.
- Valdez GR, Roberts AJ, Chan K, Davis H, Brennan M, et al. (2002) Increased ethanol self-administration and anxiety-like behavior during acute ethanol withdrawal and protracted abstinence: Regulation by corticotropin-releasing factor. Alcohol. Clin. Exp. Res 26: 1494-1501.
- 13. Heinrichs SC, Menzaghi F, Schulteis G, Koob GF, Stinus L (1995) Suppression of corticotropin-releasing factor in the amygdala attenuates aversive consequences of morphine withdrawal. Behav. Pharmacol 6: 74-80.
- Funk CK, Koob GF (2007) A CRF2 agonist administered into the central nucleus of the amygdala decreases ethanol self-administration in ethanol-dependent rats. Brain Res 1155: 172-178.
- Park PE, Schlosburg JE, Vendruscolo LF, Schulteis G, Edwards S, et al. (2015) Chronic CRF1 receptor blockade reduces heroin intake escalation and dependence-induced hyperalgesia. Addict. Biol 20: 275-284.
- O’Sullivan SJ, Malahias E, Park J, Srivastava A, Reyes BAS, et al. (2019) Single-Cell Glia and Neuron Gene Expression in the Central Amygdala in Opioid Withdrawal Suggests Inflammation With Correlated Gut Dysbiosis. Front. Neurosci 13: 665.
- O’Sullivan SJ, McIntosh-Clarke D, Park J, Vadigepalli R, Schwaber JS (2021) Single Neuronal and Glial Gene Expression in the Nucleus Tractus Solitarius in an Alcohol Withdrawal Time Series Reveals Novel Cellular Phenotypes and Networks. Research Square
- Meckel KR, Kiraly DD (2019) A potential role for the gut microbiome in substance use disorders. Psychopharmacology 236: 1513-1530.
- Yang L, et al. (2016) Systemic inflammation induces anxiety disorder through CXCL12/CXCR4 pathway. Brain. Behav. Immun 56: 352-362.
- Coller Jk, Hutchinson MR (2012) Implications of central immune signaling caused by drugs of abuse: Mechanisms, mediators and new therapeutic approaches for prediction and treatment of drug dependence. Pharmacol. Ther 134: 219-245.
- Ray LA, Bujarski S, Shoptaw S, Roche DJ, Heinzerling K, et al. (2017) Development of the Neuroimmune Modulator Ibudilast for the Treatment of Alcoholism: A Randomized, Placebo-Controlled, Human Laboratory Trial. Neuropsychopharmacology 42: 1776-1788.
- Worley MJ, Heinzerling KG, Roche DJO, Shoptaw S, Shoptaw S (2016) Ibudilast attenuates subjective effects of methamphetamine in a placebo-controlled inpatient study. Drug Alcohol Depend 162: 245-250.
- Cooper ZD, Johnson KW, Pavlicova M, Glass A, Vosbrug SK, et al. (2016) The effects of ibudilast, a glial activation inhibitor, on opioid withdrawal symptoms in opioid-dependent volunteers. Addict. Biol 21: 895-903.
- Metz VE, Jones JD, Manubay J, Sullivan MA, Mogali S, et al. (2017) Effects of Ibudilast on the Subjective, Reinforcing, and Analgesic Effects of Oxycodone in Recently Detoxified Adults with Opioid Dependence. Neuropsychopharmacology 42: 1825-1832.
- Comer S, Johnson K (2013) A Drug Candidate for Improving Opioid Analgesia and Attenuating Dependence and Tolerance: An Exploratory Trial of Ibudilast in Morphine Withdrawal and Analgesia in Heroin Addicts (P07.183). Neurology 80.
- Skosnik PD, Cortes-Briones JA (2106) Targeting the ecology within: The role of the gut–brain axis and human microbiota in drug addiction. Med. Hypotheses 93: 77-80.
- Timary PD, Leclercq S, Stärkel P, Delzenne N (2015) A dysbiotic subpopulation of alcohol-dependent subjects. Gut Microbes 6: 388-391.
- Sylvia KE, Demas GE (2018) A gut feeling: Microbiome-brain-immune interactions modulate social and affective behaviors. Hormones and Behavior 99: 41-49.
- Maniscalco JW, Rinaman L (2018) Vagal Interoceptive Modulation of Motivated Behavior. Physiology 33: 151-167.
- Schwaber J, Kapp SB, Higgins GA, Rapp PR (1982) Amygdaloid and basal forebrain direct connections with the nucleus of the solitary tract and the dorsal motor nucleus. J. Neurosci 2: 1424-1438.
- Schäfers M, Sorkin L (2008) Effect of cytokines on neuronal excitability. Neurosci. Lett 437: 188-193.
- Vezzani A, Viviani B (2015) Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 96: 70-82.
- Murphy A, Lubman DI, Mckie S, Birjal PS, Peters LA, et al. (2018) Time-dependent neuronal changes associated with craving in opioid dependence: an fMRI study. Addict. Biol 23: 1168-1178.
- Langleben DD, Ruparel K, Elman I, Pratiwadi R, Loughead J, et al. (2008) A cute effect of methadone maintenance dose on brain fMRI response to heroin-related cues. Am. J. Psychiatry 165: 390-394.
- Gorka SM, Fitzgerald DA, King AC, Phan KL (2013) Alcohol attenuates amygdala-frontal connectivity during processing social signals in heavy social drinkers: A preliminary pharmaco-fMRI study. Psychopharmacology (Berl) 229: 141-154.
- Peters S, Peper JS, Van Duijvenvoorde, ACK, Braams BR, Crone EA (2017) Amygdala-orbitofrontal connectivity predicts alcohol use two years later: a longitudinal neuroimaging study on alcohol use in adolescence. Dev. Sci 20: e12448.
- Cho Y, Crichlow G, Vermeire J, Leng L, Du X, et al. (2010) AV411 (Ibudilast) and AV1013 are non-competitive inhibitors of macrophage migration inhibitory factor: a novel induced-fit allosteric inhibition mechanism (133.11). J. Immunol 184: 133.
- Heinzerling KG, Briones M, Thames AD, Hinkin CH, Zhu T, et al. (2020) Randomized, Placebo-Controlled Trial of Targeting Neuroinflammation with Ibudilast to Treat Methamphetamine Use Disorder. J. Neuroimmune Pharmaco 15: 238-248.
- Li MJ, Briones MS, Heinzerling KG, Kalmin MM, Shoptaw SJ (2020) Ibudilast attenuates peripheral inflammatory effects of methamphetamine in patients with methamphetamine use disorder. Drug Alcohol Depend 206: 107776.
- Qureshi IS, Datta-Chaudhuri T, Tracey KJ, Pavlov VA, Chen ACH (2020) Auricular neural stimulation as a new non-invasive treatment for opioid detoxification. Bioelectron. Med 6 (2020).
- Miranda A, Taca A (2018) Neuromodulation with percutaneous electrical nerve field stimulation is associated with reduction in signs and symptoms of opioid withdrawal: A multisite, retrospective assessment. Am. J. Drug Alcohol Abuse 44: 56-63.
- Shreiner AB, Kao JY, Young VB (2015) The gut microbiome in health and in disease. Current Opinion in Gastroenterology 31: 69-75.
- Everett NA, Turner AJ, Costa PA, Baracz SJ, Cornish JL (2021) The vagus nerve mediates the suppressing effects of peripherally administered oxytocin on methamphetamine self-administration and seeking in rats. Neuropsychopharmacology 46: 297-304.
Citation: O’Sullivan SJ, (2021) The Interoceptive Antireward Pathway and Gut Dysbiosis in Addiction. J Psychiatry Depression Anxiety 7: 40
Copyright: © 2021 Sean J. O’Sullivan, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.